Nine steps for creating a master validation plan
- Kazi
- Last modified: June 1, 2024
You might wonder what a master validation plan (MVP) is and how to develop and implement one for your GMP facility.
This article can help you understand the principle of a master validation plan and what is involved in creating one.
If you are in the pharmaceutical business, regulatory authorities mandate that you develop, implement, and periodically review a master validation plan for qualifying your equipment, processes, cleaning systems, buildings, and facilities.
It is a regulatory requirement that you create one master validation plan and implement it at your site.
You need to create an effective master validation plan to ensure that your product is always safe, pure, effective, and identifiable for human use.
What should be the scope of a master validation plan?
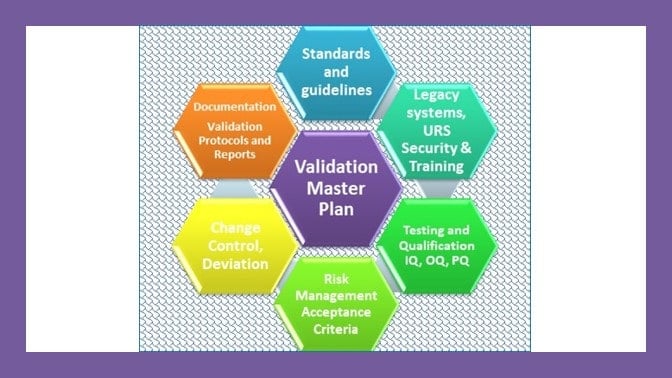
A master validation plan is a strategic document that identifies the elements to be validated, the approach for each element, the organizational responsibilities, and the documentation to be produced to ensure full consideration of product quality aspects.
The plan should show how separate validation activities are organized and inter-linked. Overall, it provides the details and relative timescales for the validation works.
The MVP must reflect all functions, departments, and manufacturing sites within the parent GMP sites or its contractors operating under GMP and GLP regulations or guidelines.
The plan applies to all existing and new drug compounds.
It covers the planning and controlling validation activities related to manufacturing drug products or active pharmaceutical ingredients (API) for clinical use or sale.
All manufacturing activities concerned with the following scenarios validation activities should be carried out following approved procedures:
1. The receipt and establishment of new Drug Products or API’s.
2. Major processing changes to existing Drug Products or API’s.
3. The construction of new manufacturing or related facilities.
4. Major alterations to existing manufacturing or related activities.
A validation master plan should be prepared for a project that consists of a range of different validation activities.
Different major projects in one facility may have a master validation plan.
Local management should plan and prepare activities, and essential documentation should be approved before starting validation activities.
240 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents included each month. All written and updated by GMP experts. Checkout sample previews. Access to exclusive content for an affordable fee.

Who is responsible for the master validation plan?
Each site should have SOPs for preparing validation documents that take account of this or are equivalent to this guideline. The Quality Assurance function should approve this documentation and the MVP.
Each site is responsible for appointing an individual responsible for validation, nominally the validation manager for a validation project.
The validation manager should be responsible for all validation activities, including the preparation of a master validation plan.
The validation manager, together with the site management staff, is responsible for ensuring SOPs are in place for preparing related validation documentation.
The master validation plan should be prepared, commented on, and approved by the senior site persons as agreed locally or nominated by a steering committee. QA approval is required.
The approval signatures on the plan, a testament to our collective effort, would normally be those of the site or manufacturing unit management team.
Master validation plans should be prepared and managed by members of the manufacturing site where the exercise is to be carried out, and personnel competent for the tasks assigned should be utilized.
Rest assured, when consultants are used to prepare plans, their work is subject to the same level of scrutiny and approval as in-house documentation, ensuring the highest standards are maintained.
In the case where third-party contractors are used to manufacture an intermediate or API, it is the responsibility of the Quality Assurance team of the lead site to ensure that the facilities, equipment, and processes at the contractor are qualified / validated in line with site requirements.
An example of computer system validation in GMP
We have analysed a list of validation activities involved during Computer System Validation in a seperate article. The post describes validation lifecycle phases, deliverables and primary elements of computer system validation.
When is a master validation plan required?
A master validation plan is needed when significant changes are made to the facilities, equipment, and processes, which may affect the quality of the product.
To determine the scope and extent of validation, a risk assessment approach should be used. The validation master plan should be available before starting any validation activity.
A new master validation plan should be prepared for projects involving major changes to existing equipment.
An MVP is not required for projects involving the installation or alteration of a single piece of equipment—these should be documented on separate validation plans and reports.
The validation master plan should be available before starting any validation activity.
Both a site MVP (for the site’s validation requirements) and project-specific MVPs may exist for some sites. The site validation committee should provide coordination as appropriate.
What should a master validation plan contain?
Each master validation plan shall describe the scope of the activities and address relevant key elements of validation affected by the change, indicating the actions and documents that will be needed.
The key elements are factors that can affect product quality.
The MVP shall identify all the components in a validation project.
Flow diagrams or matrixes can be useful as an overview and monitoring tool. A high-level process map or flowchart of the manufacturing process should also be included.
Following the issue of the master validation plan, detailed risk assessments (system and component impact assessments) should be carried out to identify items requiring qualification.
The content of the master validation plan should reflect the complexity and extent of the validation activities to be undertaken.
At a minimum, the MVP should address the following:
1. Title, statement of commitment, and approval page.
2. Summary description of the project and its scope.
3. A statement of validation policy and the objectives of the validation activity
4. References to other existing validation documents.
5. A description of the organization and responsibilities for validation
6. The validation strategy should be adopted opposite facilities and systems (i.e., process equipment and services, including automated systems), materials, quality control, and personnel, including training.
7. The intent regarding Process Validation and Cleaning Validation for each drug product range.
8. The documentation management and control system to be used.
9. A description of the validation change management process.
10. An indicative relative timescale plan.
11. Clear acceptance criteria against which the outcome of the validation exercise will be judged.
The requirements for the above should be reflected in a completed Responsibility Chart for all deliverable documentation.
A materials validation plan may be used for large projects involving many materials, but it is optional for smaller projects.
The plan or lower-tier documentation alone may cover the qualification of materials.
The validation plan should demonstrate that the validation activities have been considered and are being organized in a structured manner.
A master validation plan should be presented as a formal document suitable both as an internal guide and for scrutiny by a member of a regulatory inspectorate.
It should, therefore, be concise, easy to read, and not excessively duplicate text from documents held elsewhere in the Quality System.
Reporting requirement for master validation plan
Each validation plan should result in a report confirming that all validation activities have been completed satisfactorily.
It is recommended that a summary validation report (or master validation report) be prepared.
This report should summarize the activities undertaken, present the overall conclusions, and provide cross-references to any associated reports or follow-up actions.
Changes to the master validation plan
Members of the manufacturing site where the exercise is to be carried out should prepare and manage a master validation plan and utilize personnel competent for the tasks assigned.
Where consultants are used to prepare MVPs, their work should be subject to the same level of scrutiny and approval as in-house documentation.
Any changes to a master validation plan after the issue should be agreed upon with the project steering committee.
The MVP should not be revised once the activities described have started.
The changes should be recorded in the project documents derived from the MVP and reported in the master validation report.
Conclusion
A master validation plan (MVP) is a high-level document that outlines all validation items, prioritizes them based on risk, schedules validation activities, and documents assurances that your processes and systems function as intended.
Regulatory inspectors are eager to review your validation plans since the plan provides a detailed overview of your validation programs and activities in one document.
It is not just a suggestion, but a mandate from regulatory authorities that pharmaceutical companies develop, execute, and periodically review their master validation plan. This is to ensure that the validation and qualification of systems, processes, cleaning and testing methods, equipment, and facilities are controlled.
The master validation plan encompasses validation activities before introducing a new product or process and should address significant changes impacting them.
Before developing a master plan, it is essential to understand the various stages of the validation life cycle, including design and development, verification, qualification, commercial use, and retirement of validation items.
A typical validation workflow includes all areas under validation, such as creating individual plans and protocols, executing them, and generating reports.
Risk management is crucial for evaluating priorities, identifying processes and systems with direct versus indirect impact, and determining which to validate first. In some cases, risks can be effectively managed through in-process verification instead of comprehensive validation activities.
To avoid ambiguity, you must select your validation or verification activities through expert assessments and document all rationales in your master validation plan.
Finally, no substitute or excuse exists for not having a well-documented validation program. Regulators appreciate these records; well-managed records demonstrate your commitment to product safety.

Author: Kazi Hasan
Kazi is a seasoned pharmaceutical industry professional with over 20 years of experience specializing in production operations, quality management, and process validation.
Kazi has worked with several global pharmaceutical companies to streamline production processes, ensure product quality, and validate operations complying with international regulatory standards and best practices.
Kazi holds several pharmaceutical industry certifications including post-graduate degrees in Engineering Management and Business Administration.