A step-by-step guide to successful installation qualification (IQ)
- Kazi
- Last modified: March 13, 2025
Installation qualification (IQ) verifies if the equipment has been installed correctly according to design specifications. In other words, by IQ you will be checking if the equipment is properly installed, calibrated, and connected as per design.
Installation qualification (IQ) is one of the first testing phases during an equipment or system qualification. The other two tests are operational qualification (OQ) and performance qualifications (PQ) which are done successively after the IQ.
Do you remember the (V) shaped model commonly used to map the computer system validation life cycle? Installation qualification is the first verification step in that (V) model that is used to verify the design specification conducted earlier in the cycle.
If you are completely unfamiliar with installation qualifications, we suggest you read the article Validation in Pharmaceutical Industry first. That will give you a sense of what does installation qualification do?
In summary, the intent of installation qualification is to produce evidence that the equipment has been built and installed in compliance with their design specifications.
This article will explore what installation qualification entails, its benefits, FDA requirements, components of installation qualification, and step-by-step instructions for a successful installation qualification.
You will also know the relationship between design qualification and installation qualification, and best practices for achieving a successful installation qualification.
Table of Contents
What is installation qualification?
Installation qualification verifies and documents that all critical aspects of equipment installation adhere to design specifications and manufacturer’s recommendations.
Installation qualification (IQ) is the first phase of equipment qualification process typically performed in the pharmaceutical, biotechnology, medical devices, and other regulated industries.
Installation qualification will provide you with documented evidence that the equipment or system has been designed, developed, supplied, and installed in accordance with design drawings, the supplier’s recommendations, and in-house user requirements.
The main goal of IQ is to establish that the equipment is installed in a way that will not adversely affect its performance, reliability, and the quality of the products it produces.
Furthermore, the installation qualification test demonstrates that all the equipment’s primary features are available as designed. The equipment is one step closer to satisfactory operation.
 1")
Importance of installation qualification in regulated industries
Installation qualification is essential for guaranteeing the safety, performance, and reliability of equipment and systems in regulated industries like biotechnology, pharmaceuticals, and medical devices.
It is a methodical process that confirms and records that every important part of an installation complies with the standards.
Your company can reduce risks, avoid expensive mistakes, and preserve the integrity of its processes by undertaking installation qualifications.
In industries where the quality and safety of products are top priorities, installation qualification is particularly critical.
A defective installation of manufacturing equipment or a cleanroom, for instance, can have major repercussions on pharmaceutical products, including contamination, faulty products, market recalls, and other regulatory non-compliance.
Installation qualification is therefore more than simply a good idea—it is a legal requirement that you must be strictly adhered to.
When you will conduct an installation qualification successfully you can be benefitted by:
i. Ensuring that the equipment is installed correctly and functions as intended, minimizing the risk of equipment-related issues that could have impacted product quality.
ii. Helping your company meet the regulatory requirements set by organizations like the FDA, EMA, and other relevant authorities.
iii. Identifying and mitigating potential risks associated with equipment installation, preventing costly failures and downtime.
iv. Providing a comprehensive and detailed record of the installation process, and facilitating future audits and inspections.
v. Improving productivity since correctly installed equipment is more likely to operate efficiently, and reduced operational costs.
250 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents are included each month. All written and updated by GMP experts. Check out sample previews. Access to exclusive content for an affordable fee.
Regulatory requirements for installation qualification
You must conduct Installation qualifications in compliance with the rules and regulations developed by regulatory agencies like the Food and Drug Administration (FDA) and the European Medicines Agency (EMA).
Regulatory Agencies have provided guidelines; qualification outlines together with the minimum standards to maintain. You must follow these guidelines and reporting requirements during installation qualification at the minimum.
Regulatory requirements require you to create and adhere to a validation master plan that describes the strategy, obligations, and processes for installation qualification.
Similarly, the EMA’s Good Manufacturing Practice (GMP) guidelines emphasize the importance of installation qualification in ensuring the quality and safety of pharmaceutical products.
These guidelines require companies to perform installation qualification for critical equipment and systems and to document the results in a comprehensive manner.
Relationship between design qualification (DQ) and installation qualification (IQ)
Both design qualification (DQ) and installation qualification (IQ) are interrelated.
DQ ensures that equipment is designed to meet specific requirements, while IQ verifies that the equipment is correctly installed to those requirements.
Design qualifications are typically outsourced to specialized firms. Developers and IT/Engineering develop the required design specifications.
Design specifications shall specify minimum requirements for system hardware and software, or ancillary software tools and the baseline configuration. This stage identifies how the system will meet the functional specification.
On the other hand, Installation qualification (IQ) verifies and documents that all critical aspects of equipment installation adhere to predetermined design specifications and manufacturer’s recommendations.
Both DQ and IQ are essential steps in the equipment qualification process, ensuring that the equipment meets its intended purpose.
When is equipment qualification necessary?
Equipment used in pharmaceutical manufacturing processes can range from basic balances and scales to sophisticated and automated process control and monitoring systems.
Since the equipment is so different, it is not practical for you to use a single set of principles to support the same level of qualification program, including operational qualification. It would also be incorrect from a scientific standpoint.
You should conduct a risk-based categorization of all equipment and systems and list them between,
– Non-Critical
– Commercial off the shelf (COTS)
– Critical, and
– Bespoke
You should follow the GAMP (Good Automated Manufacturing Practice) categorization standards created by the International Society for Pharmaceutical Engineering (ISPE) when determining which equipment needs qualification testing.
GAMP offers a framework for validating automated systems based on equipment criticality. Software and hardware are categorized by GAMP according to their complexity, which aids in figuring out how much qualification work is necessary.
GAMP divides equipment and systems into five categories:
Category 1: Infrastructure Software
These are fundamental computerized systems, such as network infrastructure, database management systems, and operating systems.
Usually, these elements have less of an effect on the final product’s quality and patient security. These are therefore viewed as non-critical.
Category 2: Non-configurable Software
Non-configurable software includes commercial off-the-shelf (COTS) applications that are used without any customization or modification.
For example, standard office software like word processors and spreadsheet applications.
While GAMP 2 systems are not directly involved in pharmaceutical processes, they may handle data that needs to be validated.
Category 3: Configurable Software
Configurable software is customizable to some extent but does not require full software development.
Laboratory information management systems (LIMS) and data acquisition systems are the best examples.
Category 4: Bespoke Software
Bespoke software refers to custom-developed software specifically designed to meet unique requirements and processes within a pharmaceutical operation.
These systems are often critical to product quality and safety and require more extensive validation efforts.
Category 5: Process Control Systems
Process control systems are complex systems responsible for controlling and automating manufacturing processes in pharmaceutical facilities.
These systems have a direct impact on product quality and safety and are considered the most critical and complex to validate.
Follow the diagram below to make a decision on the extent of equipment qualification activities that are necessary based on the product risk and criticality of equipment operation on products.
 2")
A good idea is to review the GAMP categorization and assess the impact of equipment on product quality. Then classify all of your equipment and systems. This will help you make an informed decision on how many qualification activities will be necessary.
250 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents are included each month. All written and updated by GMP experts. Check out sample previews. Access to exclusive content for an affordable fee.
What are some installation qualification examples?
Here are some common examples of equipment that you may or may not have to undertake installation qualification depending on their criticality and complexity:
– Laboratory Glassware (Beakers, Flasks, Pipettes)
– Magnetic Stirrers
– Hot Plates
– pH Test Strips
– Safety Equipment (Laboratory Coats, Safety Glasses)
– General Purpose Centrifuges
– Magnetic Separators
– Vortex Mixers
– Laboratory Shakers
– Water Baths
– Laboratory Incubators
– Laboratory Freezers (Non-critical storage)
– General Purpose Ovens
– General Utility Carts
– Laminar Flow Hoods (for non-sterile applications)
– Vacuum Pumps (for non-critical applications)
– Non-Sterile Sample Containers
– Standard Laboratory Balances (Non-analytical balances)
– Balances and Scales
– Autoclaves
– Incubators
– Ovens
– pH Meters
– Conductivity Meters
– HPLC Systems
– UV-Visible Spectrophotometers
– Dissolution Testing Apparatus
– Particle Size Analyzers
– Freeze Dryers (Lyophilizers)
– Water Purification Systems
– Tablet Press Machines
– Homogenizers
– Refrigerators
– Freezers
– Environmental Monitoring Systems
– Automatic Capsule Filling Machines
– High-Performance Liquid Chromatography (HPLC) Systems (Critical for analytical testing and product quality assessment).
– Gas Chromatography (GC) Systems (Critical for analyzing volatile compounds).
– Mass Spectrometers (Critical for molecular identification and structural analysis).
– Spectrophotometers (UV-Vis, IR, Fluorescence) (Critical for quantitative analysis and drug release testing).
– Particle Counters (Critical for monitoring and controlling cleanroom environments).
– Aseptic Filling Machines (Critical for sterile product filling to maintain product integrity and patient safety).
– Lyophilizers (Critical for freeze-drying temperature-sensitive pharmaceutical products).
– Sterilization Autoclaves (Critical for ensuring aseptic conditions and product safety).
– Disintegration Testers (Critical for assessing the disintegration properties of solid dosage forms).
– High-Speed Tablet/Capsule Counting Machines (Critical for accurate counting and packaging of tablets and capsules).
Bespoke systems:
– Customized Mixing and Blending Systems (Designed for specific formulations or challenging mixing requirements).
– Custom-built Reactors and Fermenters (For specialized pharmaceutical synthesis and bioprocessing).
– Custom Analytical Instruments (Developed for unique analytical methods or specific sample types).
– Custom-Designed Cleanrooms and Isolators (For specialized aseptic manufacturing and containment).
– Automated Robotic Systems for Sample Handling (Designed to automate complex sample preparation and handling).
– Specialized Microscopes (For unique inspection and analysis tasks in pharmaceutical research).
– Custom Temperature and Humidity Chambers (For stability testing under specific conditions).
– Process Analytical Technology (PAT) Systems (Customized for real-time monitoring and control of manufacturing processes).
– Customized Dissolution Testing Apparatus (Tailored for specific dosage forms or release rate requirements).
– Process Control and Monitoring Systems (Designed for specific manufacturing processes and data collection needs).
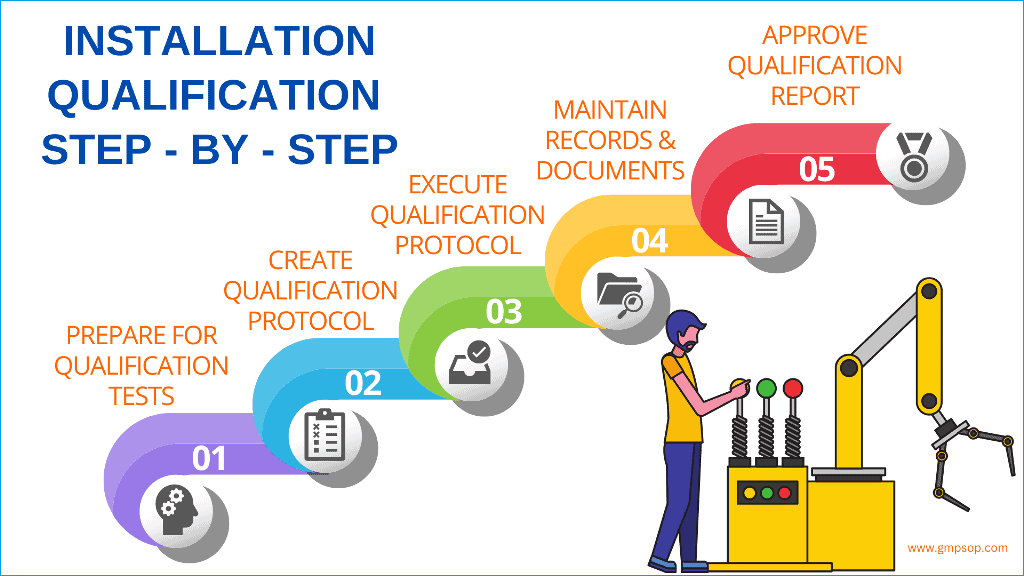
How to complete installation qualification, step by step
Installation qualification is a process. You can break it down into five different steps to complete one successfully.
Step 1: Prepare for the installation qualification
It is critical for you to prepare the installation qualification plan prior to commencing the tests.
You should understand the objective of the installation. Define the important parts of the equipment or system that will be verified and ensure that all relevant resources are accessible.
First, you should establish the installation qualification’s scope. This includes identifying the equipment or system being verified, as well as any auxiliary components or services.
You should ensure that all key issues are handled during the qualifying process by explicitly specifying how to deal with each issue.
Following that, it is critical to identify and evaluate the risks involved with the installation. This requires carrying out a risk assessment to identify potential risks, estimate their severity and likelihood, and devise appropriate risk mitigation measures.
250 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents are included each month. All written and updated by GMP experts. Check out sample previews. Access to exclusive content for an affordable fee.
Step 2: Create an installation qualification protocol
After you prepare for the qualification, the next step is to develop an installation qualification protocol.
The protocol should describe the equipment or system, tests to complete, test methodology, acceptance criteria, test results, the person responsible to complete the tests, dates, and initial.
The installation qualification protocol should include a full explanation of the qualification process to be carried out, including specific tests or inspections. It should also specify the acceptance and rejection criteria for the test, as well as the documentation requirements.
The protocol should explicitly describe the roles and responsibilities of the people who will be participating in the qualification process. The protocol should identify qualified persons who will perform the test as well as those who will assess and approve the results.
Also, you should include references to the documents to be used, deviation handling, and change management procedure in the protocol.
Step 3: Execute the installation qualification protocol
During the execution phase, the qualified personnel will perform the specified tests and inspections to ensure that the installation meets the predefined acceptance criteria.
This will include verifying the correct installation of equipment, checking the calibration of instruments, and assessing the functionality of control systems.
It is critical to follow the qualification protocol meticulously and document the results accurately.
Any deviations or non-conformities should be recorded and addressed appropriately.
By adhering to the protocol and documenting the results systematically, you can ensure the integrity and traceability of the qualification process.
Step 4: Keep records of the installation qualification process
Installation qualification relies heavily on documentation. It acts as a record of qualification actions, offers evidence of compliance, and makes future audits or inspections easier.
You must record all important information, including test data, observations, conclusions, etc. in a clear and organized manner throughout the documentation phase.
Documenting the installation details, the qualification strategy, the qualification protocol, and any deviations or non-conformities detected during the process are all part of this.
It is critical that the documentation be thorough, accurate, and easily available. Maintaining effective version control, employing standardized templates and formats, and storing documentation in a secure and centralized location are all part of this.
You should keep in mind that proper documentation is critical for demonstrating compliance, facilitating knowledge transfer, and allowing for successful troubleshooting or maintenance in the workplace.
Step 5: Review and approve the installation qualification report
The final step of the installation qualification process is to review and approve all results and supporting records generated during the qualification steps.
The qualified personnel should examine the qualification findings comprehensively to confirm if all requirements have been met and if the installation is suitable for its intended function.
The personnel in charge of approving the qualification report should carefully study the documentation, including the test data, observations, and conclusions.
They should ensure that all tests and inspections were carried out correctly, that the acceptance criteria were met, and that any deviations or non-conformities were resolved effectively.
The authorized personnel must approve the report using wet signatures or electronic approvals to confirm that the installation qualification has been finalized.
What are the essential components of installation qualification protocol?
When you will devise an installation qualification protocol it is a common practice to include operational qualification tests with it.
That will provide you with an upper hand in merging the tests, troubleshooting, and final reporting of the verifications.
Following are the key components of an installation and operational qualification that you should use when building your protocol:
1. Include project details such as:
– Project name
– Project number
– Equipment
– Serial Number
– Manufacturer
– Model Number
– Process Line/Location
– Protocol number
2. Add the objective of the installation qualification protocol
Write the objective of the protocol defining the installation qualification (IQ) and operational qualification (OQ) requirements and acceptance criteria for the equipment with location i.e., packaging or manufacturing, and the facility.
Include a brief description of why these qualifications (IQ/OQ) are required i.e., new equipment.
Provide a statement like successful completion of this protocol will provide a high degree of assurance that the equipment has been installed and operates in accordance with the site requirements, specifications, and manufacturers recommendations and is in compliance with cGMP and site policies.
3. Develop the scope of the installation qualification protocol
Create the scope of the protocol by including equipment or system name. Include all sub-systems that are part of the main system. If there is a validation protocol provide a reference of the protocol no.
Include the other equipment or system within the same area that are not part of the protocol. If applicable, reference the document number in which they are validated e.g. cleaning validation number.
4. Provide a description of the equipment or system
Provide a clear and precise description of the equipment and how it operates. This should be limited to one or two paragraphs and should focus on the GMP and key installation and operational characteristics of the equipment.
The information may be available in a manual but it may need to be tailored to the protocol.
5. Provide installation qualification methodology to be used
State how the validation study will meet site and cGMP requirements.
Include the reference to the risk assessment that was conducted. Detail how would the qualification study will target the risk areas that are considered to pose a high risk to product safety, efficacy, purity, and identification.
Now list the tests that will be performed as part of the protocol such as verification of installed equipment.
6. Develop the acceptance criteria against which the results will be measured
You have to set acceptance criteria against each test and inspection that appear on each test script. The equipment will be deemed to be qualified when all of the following conditions are true:
– All activities listed in the check sheets have been performed.
– The equipment or system installation and operation have conformed to the acceptance criteria for each test.
– All entries of data have been signed and dated by the person or persons who did the work.
– All tests have been reviewed
– Any deviations that have occurred during the qualification have been satisfactorily resolved or accepted and have been approved.
250 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents are included each month. All written and updated by GMP experts. Check out sample previews. Access to exclusive content for an affordable fee.
7. Add references to the documents that are used to develop installation qualification protocol
Insert all document that is relevant e.g. SOPs, Previous qualification documents if this is a requalification, standards such as standards, etc.
8. Identify the stakeholder’s responsibility and authority during tests
Clearly identify and assign responsibilities to the Validation team, Engineering, Production, Quality Assurance, and Regulatory Compliance teams. Identify who has the independent authority to finally assess the results and approve the protocol.
9. Describe how the deviations will be handled during installation qualification?
Unplanned deviations from the protocol are common during qualification studies.
You should prepare a deviation log to list all deviations from the approved methodology, procedure, or expected versus actual results.
Keep records of detailed investigations, summary of assessments, and final corrective actions. Show how those deviations may or may not have impacted the overall outcome of the installation qualification study.
The installation and operational qualification cannot be considered valid for use until all critical deviations have been resolved. All deviations must be resolved or justified prior to protocol completion.
10. Add a section for change control and re-validation
If you will experience a change to the protocol you have to deal with proper change management procedure. All changes to the procedure must be assessed by the authorized person and approved before the amendments can be made.
11. Include appendices in the protocol
Include appendices one for verification test scripts and acceptance criteria for each equipment, component, or system that are under test within the scope of the protocol.
In each appendix include the test number, equipment number and description, installation date, acceptance criteria, test results, test completed by name and signature.
12. Signature identifier
Include a signature identifier log where each person has participated in the installation qualification is identified by printed name, company/position, and corresponding signature and written initials.
250 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents are included each month. All written and updated by GMP experts. Check out sample previews. Access to exclusive content for an affordable fee.
 4")
What is included in the installation qualification report?
After you complete the verifications of installation qualification, you have to compile all your findings and observations into an installation and operational qualification report.
The report should include:
i. Scope and objectives: A clear description of the goals the installation qualification protocol has achieved.
ii. Equipment or system description: State the equipment or system name, sub-parts, location, serial number, make, and model number which are within the scope of the qualification process.
iii. Qualification test details: Comprehensive documentation of the testing methodologies you have taken, including any deviations encountered and how they were addressed.
iv. Test results: Detailed results of all tests performed during the qualification process. Ensure to include the predetermined acceptance and rejection criteria. Make comments if those have passed or failed the criteria.
v. Documentation and training: Include the details of all documentation referenced in the protocol. Describe if you have to undertake training. Keep the records for all training for employees.
vi. Conclusion: A final assessment of whether the equipment installation was successful and met the acceptance criteria.
Troubleshooting common issues during installation qualification
Most people who have experience in installation qualification would admit that it is a challenging task. Challenges include inadequate preparation, poor documentation, insufficient resource allocation, and lack of clear communication.
If you would fail to properly define the scope, identify all risks and mitigations, set inaccurate acceptance criteria, or assign tasks to incompetent personnel, the effort will lead to potential issues down the line.
It is important to invest time and effort in the preparation phase to ensure a successful qualification process.
Some common issues encountered during installation qualification include:
i. Equipment Malfunction: Stop the testing. Address any issues with equipment functionality immediately and perform corrective actions as necessary.
ii. Documentation Errors: Verify that all documentation accurately reflects the installation process, design specification and make corrections if needed.
iii. Calibration Problems: Ensure that all testing equipment is calibrated correctly to prevent bias or inaccurate results.
Best practices for successful installation qualification
Follow these best practices to ensure a successful installation qualification:
i. Thorough Planning: Develop a comprehensive qualification protocol before starting the installation process.
ii. Attention to Detail: Pay close attention to the manufacturer’s instructions and follow them meticulously.
iii. Well-documented processes: Maintain detailed records throughout the qualification process.
iv. Proper Training: Ensure that personnel involved in the qualification process are adequately trained and qualified.
v. Validation of testing methods: Validate all testing methods used during the qualification process to ensure the accuracy and reliability of results.
250 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents are included each month. All written and updated by GMP experts. Check out sample previews. Access to exclusive content for an affordable fee.
Conclusion
Installation qualification is a vital step in ensuring that equipment and systems are correctly installed and functioning as intended.
By following the step-by-step guide and adhering to best practices, you can achieve successful installation qualification, reduce risks, and ensure compliance with regulatory requirements for your business.
The development of installation qualification protocol is of the utmost important task for your success. The essential elements of creating the protocol that we have detailed in this document are the best industry practices.
Properly qualified equipment is critical for producing high-quality products and maintaining the overall efficiency of operations in regulated industries.
If you would like to know more on how to prepare acceptance criteria to use using verification of common pharmaceuticals equipment please read our article on operational qualification.
FAQ - installation qualification (IQ) process
1. What is Installation Qualification (IQ)?
Installation qualification (IQ) is the first of the three testing phases of equipment qualification in regulated industries. It precedes the other two tests operational qualification (OQ) and performance qualification (PQ).
IQ ensures that equipment is installed correctly, adhering to predetermined design specifications and the manufacturer’s recommendations.
2. What is Installation Qualification (IQ)?
Installation Qualification (IQ) is the initial phase of equipment qualification in regulated industries, such as pharmaceuticals and medical devices. It ensures that equipment is installed correctly, adhering to predetermined specifications and the manufacturer’s recommendations.
3. Why is Installation Qualification important?
IQ is essential for several reasons:
It ensures equipment functions as intended, minimizing the risk of issues affecting product quality.
It helps companies comply with regulatory requirements, particularly those set by the FDA and other relevant authorities.
IQ reduces the risk of equipment-related failures and downtime, leading to increased efficiency and cost savings.
4. What is the relationship between Design Qualification (DQ) and Installation Qualification (IQ)?
DQ ensures that equipment is designed to meet specific requirements, while IQ verifies that the equipment is correctly installed according to those requirements. Both DQ and IQ are crucial steps in the equipment qualification process.
5. What are some examples of equipment that undergo Installation Qualification?
Various types of equipment undergo IQ, including manufacturing machinery, laboratory instruments, computerized system software and hardware, HVAC systems, and utilities like purified water systems.
6. What are the regulatory requirements for Installation Qualification?
Installation qualification must be conducted in compliance with the rules and regulations developed by regulatory agencies such as the FDA and the European Medicines Agency (EMA). Companies must create and adhere to a validation master plan and document the entire qualification process.
7. What are the essential components of an Installation Qualification Protocol?
An IQ protocol should include project details, the objective of the qualification, the scope of the protocol, installation qualification methodology, acceptance criteria, references to relevant documents, stakeholder responsibilities, deviation handling procedures, change control and re-validation processes, and appendices for test scripts and signatures.
8. How do you complete Installation Qualification, step by step?
The steps for a successful IQ process include preparing for the qualification, creating an IQ protocol, executing the protocol, keeping records, reviewing and approving the qualification report, and addressing any deviations that may occur during the process.
9. What is included in the Installation Qualification report?
The Installation Qualification report should include the scope and objectives of the qualification, equipment or system description, details of the qualification tests, test results with acceptance criteria, documentation and training information, conclusions, and any identified troubleshooting issues.
10. What are some common issues encountered during Installation Qualification?
Common issues during IQ include equipment malfunction, documentation errors, calibration problems, and inadequate preparation. Proper planning and attention to detail are crucial to avoid such issues.
11. What are the best practices for successful Installation Qualification?
Key best practices include thorough planning, attention to detail in following manufacturer’s instructions, well-documented processes, proper training for personnel, validation of testing methods, and addressing deviations effectively.
 5")
Author: Kazi Hasan
Kazi is a seasoned pharmaceutical industry professional with over 20 years of experience specializing in production operations, quality management, and process validation.
Kazi has worked with several global pharmaceutical companies to streamline production processes, ensure product quality, and validate operations complying with international regulatory standards and best practices.
Kazi holds several pharmaceutical industry certifications including post-graduate degrees in Engineering Management and Business Administration.