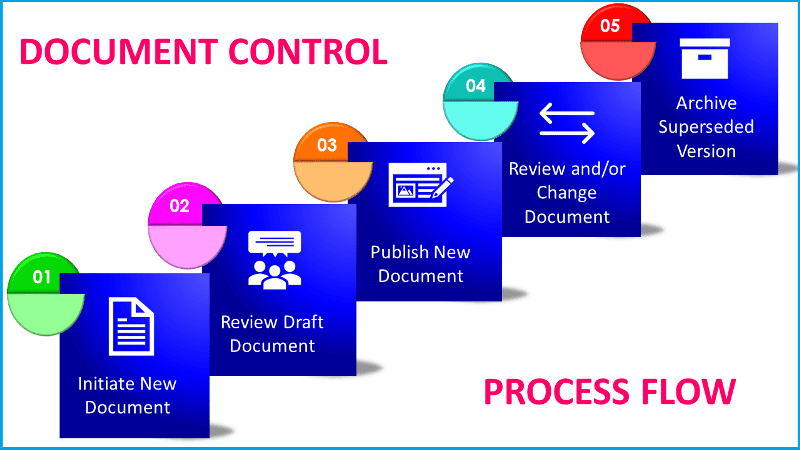
Document Change Control Process in GMP Environment
- Kazi
- Last modified: April 2, 2024
Purpose
In this article we will detail the steps and mechanisms inovolve in document change control process which is important to maintain a good Quality Management System (QMS) within a GMP controlled environment.
Scope
This article covers the controls in place for document generation and numbering, review and approval, document change control, internal and external distribution, and the Document Control Registers. All documentation being generated with a view to being controlled must be raised, reviewed, approved and controlled as per the requirements of national regulatory body and international guidelines. It also identifies the Good Documentation Practices (GDP) that must be implemented by all staff.
Document Generation and Approval
Document Numbering
Quality Procedures, Work Instructions, Quality Forms and Test Record Sheets must all be identified with a unique number prior to issue. The Quality Management System utilises a numbering system that collates all the related departmental procedures together and also clearly identifies where a form is directly connected to a specific process and procedure.
The prefixes used to identify departmental documents are as follows:
(Different organisation adopt varying approaches while designing the document identifier. There is no rule or guidance mandated in this case except the idea is to maintain the traceability at any given process.)
MGT – Corporate Management
RQA – Regulatory Affairs and Quality Assurance
CSV – Customer Service
TSV – Technical Service
LGS – Logistics
FIN – Finance
SAM – Sales and Marketing
Example:
The first Quality Procedure covering a Customer Service process will be identified as CSV 1-1. Any forms raised in support of this particular process will be identified as CSV 1-1A, B, C and so on.
A subsequent procedure for another process will be identified as CSV 1-2, with supporting forms being CSV 1-2A, B, C and so on.
The Regulatory Affairs/QA Department is responsible for allocating numbers to documents which are to be controlled in the Quality Management System. The numbers are taken from a register under their control.
210 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents included each month. All written and updated by GMP experts. Checkout sample previews. Access to exclusive content for an affordable fee.
Document Generation
All controlled documents are to be created using MS Word, MS Excel or MS Visio and formatted to match existing documents in the same series. For ease of viewing, any MS Visio documents will be printed and scanned, prior to placing the scanned image within the Quality Management System documentation on the organisation’s network.
Any staff may generate a draft document for consideration for introduction into the Quality Management System. If deemed acceptable by the Quality Manager, format and content changes may be made prior to formal issue and use, to ensure correct compliance to requirements.
Document Approval
Controlled Quality Procedures must be generated, reviewed and approved for adequacy by individuals who have either management responsibility or a thorough working knowledge of the process concerned. The individual reviewing the document for accuracy of content must not be the originator, but the approval signatory, indicating the completed document may be released for issue into the QMS, may be the originator in cases where the document has been originated by a Quality authority.
The review and approval boxes on such documents have the signatories’ names in print, their signature and date, with the current issue status of the document appearing in the footer. Also shown is the Mandatory Review Date, which is a nominated date based on a period not exceeding two years since the last revision of the document. Procedures are flagged for review in this manner to ensure their content is still current.
Document Control Registers
Under the control of the Quality department are a series of company registers which control:
– The allocation of new QMS document numbers
– The issue and control of Document Change Request forms
– Records of controlled document distribution – Internal and External
– Records of externally generated/supplied documents
Document Change Control
Any amendments made to a controlled document within the QMS must be proposed, reviewed and approved via the formal Document Change Control mechanisms. Central to this process in the Document Change Request (DCR) form. The Change Control process steps include.
Raising the DCR
Any staff member is in a position to generate a DCR to propose changes to a document. When requested, the Quality department will provide the originator with a blank form and allocate the next sequential number from the DCR Register.
Completing the DCR
The originator completes initial part of the form, giving full details of the document to be changed, the changes that are being proposed, the reasons for the change and attaches a marked up copy if applicable.
Where the change could affect another controlled document, details must be given on the DCR.
The originator then circulates the DCR to the nominated departmental reviewers, who review and approve the proposed changes if considered acceptable.
Note: The circulation list of nominated reviewers must, wherever possible, be the original approval authorities, or individuals with sufficient knowledge of the process. These individuals are generally identified by the Quality department at the time of issuing the blank DCR. This list will be the minimum reviewers who are considered to be stakeholders in the process affected by the proposed changes.
The originator may choose to involve additional reviewers to those nominated, but must ensure that the form is fully completed and signed by the initial nominated reviewers.
Note: For some Quality documents it is permissible for the changes to be reviewed only by the QUALITY department, as no other stakeholders will have the appropriate appreciation of the content matter.
Review Considerations
Each reviewer must consider the potential impacts that the proposed change may have to processes under their control or the potential impacts on other areas of the business. The Change Control form should designed to enforce a Risk Management approach to the review and approval of any changes being made to company controlled documentation.
The reviewer’s comments and signatures are recorded in second part of the DCR.
Where the proposed change is not considered acceptable, the applicable reviewer must liaise with the originator to discuss and negotiate amendments. It is the responsibility of the originator to re-circulate the amended change proposal to reviewers who have already accepted the previous version of the change. All parties must be in agreeance before the change can be processed.
Processing the Accepted Change
Once the reviewer’s acceptance signatures have been obtained, the originator submits the DCR to the Quality department for processing. Quality then arrange for the changes to be made to the controlled document within the QMS, superseding both soft copy and hard copy of the old issue document.
The new issue of the document is released onto the QMS and a new hard copy master placed in the QMS file. The QMS hard copy will be stamped ‘Control Copy’. Note: The Soft copy of all QMS documents is considered the Master Copy for retention purposes.
If applicable, copies of the new issue document will be internally or externally distributed to recipients of the previous issue. As each of these activities is undertaken, the actions are recorded in Part 3 of the DCR form. When all actions are complete, the DCR form is closed and the DCR register amended accordingly.
Product Changes Affecting Regulatory Requirements
Where information is received from the external Manufacturers in relation to a change in the design, manufacture, test, packaging, or labelling of a product, the details are assessed by the Quality department to determine if the change affects the registration of the device with the Regulatory Bodies.
The result of such change assessment is annotated on the applicable Change Notice, signed by the reviewing Quality representative and filed in chronological order in the Completed Change Notices in the Quality department. Changes affecting product performance or packaging are forwarded to the Product Manager for discussion with customers if applicable.
Control of Document Distribution
The current versions of all QMS documents should be available to all staff on the company network. Any document printed off are considered “Uncontrolled Copies” unless stamped in accordance with this procedure. Uncontrolled documents must not be used for any process, purpose or activity where the product safety, quality or reliability may be adversely compromised.
The use of such documents should be considered a performance issue.
Any distributed copies of controlled documents must be identified and recorded via the following Document Distribution system. These activities are the responsibility of the Quality department.
The Quality department photocopy the required quantity of the document to be distributed
The front page of the document is stamped with the relevant copy number for the recipient. The copy number is determined by referencing the key within the Document Distribution Register
– If the document is for Internal distribution, Quality raise and complete the Distribution Form allocating a unique reference number from the Distribution Register.
– If the document is for External distribution, Quality raise and complete the Transmittal Form allocating a unique reference number from the Transmittal Distribution Register
– The details are entered into the Document Distribution database
– Internally distributed documents are distributed and the recipient signs and returns the Distribution form
– Externally distributed documents are posted to the recipient, who completes and returns the Transmittal form
– Where applicable, the recipient must return any superseded copies of the document to the Quality department
– Quality updates the Document Distribution Database and file completed Distribution/Transmittal Forms
In cases where recipients have not returned the signed and completed Distribution or transmittal forms in a timely manner, nominally 7 days for Internal and 14 days for External distribution, the Quality department will contact the recipient to expedite a response.
Control of External Documents
In some cases company receives copies of documents provided by an external sources. These documents also have a level of control applied to them.
Typically such documents will fall into two categories:
– Controlled externally distributed documents from a subcontractor/supplier – Arrangement must be made to ensure these documents to be automatically re-issued to recipient organisation if the document is amended.
– Unmaintained externally generated documents such as International Standards – These documents will not have the issue status automatically maintained)
Controlled External Documents
The Quality department maintains a register of controlled documents which have been received into the organisation. The register is available within the Document Control Databases on the company network.
Typically, such documents are copies of quality procedures and work instructions in support of processes where the recipient organisation interface with a subcontractor or supplier. Copies of these documents can be distributed within recipient organisation upon request and will be controlled and distributed as per section above.
Control of External Standards/Unmaintained External Documents
The Quality department is responsible for registering all copies of External Standards and other unmaintained external documents which are available to staff for reference purposes. The Master List for these documents is available within the Document Control Databases on the company network.
Predominantly these documents are International Standards and cannot be considered to be at the latest issue status as they are not formally maintained. The information within them is not to be used for any purpose, activity or process which may affect product safety, quality or reliability, unless the issue status has been verified as current by the Quality department.
These documents may be held in either Hard or Soft copy format by the Quality Department.
Good Documentation Practices
Standardizing Good Documentation Practices
G MP sites must have a standard approach to the correct protocol when completing, processing and controlling documentation.
The preferred protocol is based on Industry Best Practises and satisfies the expectations and legal requirements.
Making Hand Notations on Documents
When completing Forms or Handwriting Information on documents, the following is to apply:-
– All entries are to be in ink – Pencil is not permitted
– Every Box on any form must have an entry of some description.
– Boxes which are not applicable must be marked N/A or be struck through with a diagonal line.
– This demonstrates that the aspect has been considered, rather than overlooked.
– Authorisation of documents should always be indicated with: Name (in Print), Signature and Date
Correct Date Format
To reduce the risk of misreading dates on documents, staffs are to use one date format consistently (ie, DD/MMM/YYYY)
When working with documents received from other affiliate sites, staff should ensure they establish the correct date, as different countries different dating style.
Hand Amendments & Errors
It is particularly important to follow strict protocols shown below when making Hand Amendments to documents:
– NO use of Liquid Paper or White Out Tape whatsoever.
– Do Not scribble out errors or obliterate them
– Amend errors with a Single Strike Through, and then Initial or Sign and Date the amendment.
– The information entered in error Must Always be Visible if required to be scrutinised by a third party at some future date.
– If the quantity of errors is significant, and it is acceptable to do so, then a fresh Form/Document should be raised and re-completed correctly.
Photocopying and Document Distribution
Staff wishing to print copies of documents from the QMS Must stamp the hard copies with “Uncontrolled Copy” stamp – (Stamp held By Quality).
Such documents Must Not be used in support of processes which may affect the quality and accuracy of the Product or any of the Services we provide. The content cannot be considered current once printed from the QMS.
Staff may request Controlled Copies of any QMS documents, which will be stamped and formally issued by Quality to a named individual. This ensures holders automatically receive updated versions of all documents they hold.
The above requirements also apply to documents located on other organisation’s
Quality Records
All documentation generated in relation to the Document Control activities within this procedure are to be considered Quality Records, and are to be controlled and archived as per established Quality Records management procedure.

Author: Kazi Hasan
Kazi is a seasoned pharmaceutical industry professional with over 20 years of experience specializing in production operations, quality management, and process validation.
Kazi has worked with several global pharmaceutical companies to streamline production processes, ensure product quality, and validate operations complying with international regulatory standards and best practices.
Kazi holds several pharmaceutical industry certifications including post-graduate degrees in Engineering Management and Business Administration.
Keep all the articles coming. I love reading your posts. All the best. Dillon Holness