How to develop GMP compliant software for quality management
- Kazi
- Last modified: March 13, 2025
It has long been identified that if a pharmaceutical manufacturing business can automate its quality management systems using validated and reliable GMP compliant software applications, its productivity and compliance can improve in numerous ways.
A GMP compliant software system can effectively replace conventional paper-based approaches, leading to significant time and cost savings for the business. With today’s AI-driven technology, the requirements for GMP compliant software have become more prevalent, as has the complexity of regulations.
Governments highly regulate pharmaceutical industries. They must follow stringent procedures to validate and maintain their information management systems for GMP compliance. For example, companies located in the USA must comply with the USFDA 21 CFR part 11 regulations.
Businesses must create detailed user specifications that support their system design to comply with regulatory requirements for software systems. The biggest challenge is building applications (in-house or outsourced) that reflect their operational designs and remain compliant with those regulations.
As a result, businesses can incur significant costs from employing experienced business analysts and developers to build automated systems. It also takes a considerable period to develop a software system that works.
This post will explore the essential requirements of a software system, which may guide you in developing an application that is reliable, user-friendly, fit for purpose, and compliant with GMP and other regulatory requirements.
The scope of this post is to focus on building a software application that can be used for automating quality concern investigation processes within the quality management area, such as:
i. Product complaint investigation management.
ii. Deviation investigation management.
iii. Change control management.
iv. Laboratory out-of-specification investigation management.
v. Corrective and preventative actions.
For demonstration purposes, I have chosen the regulatory requirements of USFDA 21 CFR part 11.
Table of Contents
Key Takeaways
- Transitioning from paper-based systems to GMP compliant software can significantly enhance the productivity and compliance of quality management systems.
- GMP software used in the pharmaceutical industry must comply with regulatory standards such as USFDA 21 CFR Part 11, which governs electronic records and signature management during investigations. Compliance with these regulations will help you achieve high reliability, accuracy, and integrity in your quality assurance processes.
- Effective GMP compliant software should support automation, improve data accuracy, maintain the integrity of electronic records, robust reporting and analytical capabilities.
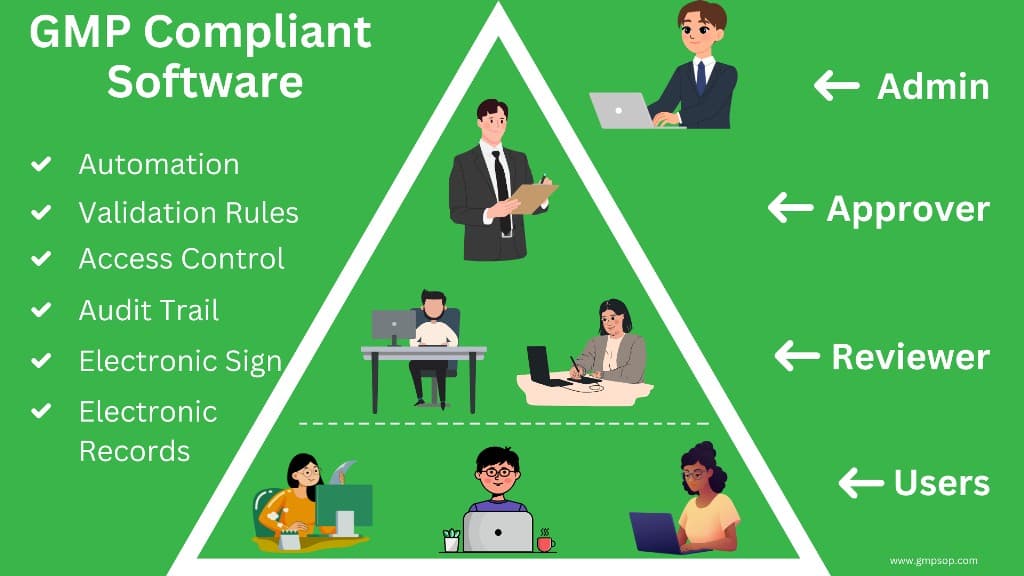
- Essential functionalities of GMP compliant software include electronic signatures, audit trails, strict user access controls, modular designs for specific investigations, and comprehensive database management for secure record storage and retrieval.
- The software should include distinct modules for quality assurance investigations, such as product complaints, deviations, change controls, laboratory investigations, and CAPA. Each module should be designed to allow independent operation but integrate with a centralized database.
- To meet GMP requirements, the software must implement validation rules that ensure chronological activities are logged and workflow processes are logical. The software should also be able to check user errors and guide users to follow validation rules.
- A hierarchical user system (entry-level, reviewer, approver, and administrator) must be established to ensure accountability and restrict user access based on responsibilities, preserving the integrity of records.
- Robust security measures, including encrypted logins, automated backups, and contingency planning for disaster recovery, are critical to protect electronic records and ensure system reliability.
- The software should be developed to generate insightful reports on closed and open investigations, CAPA actions, quality trends, and other compliance metrics, supporting your organization's effective decision-making and governance.
- The software system must be intuitive to users, with accessible help files, automated installation processes, and cross-platform compatibility. This will ensure the application's user-friendly operation.
250 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents are included each month. All written and updated by GMP experts. Check out sample previews. Access to exclusive content for an affordable fee.
Fundamental requirements of GMP compliant software
The critical objectives for developing a quality management software application are to make the quality concern investigations processes faster, more reliable, accurate, secure, and permanent for future reference.
A carefully designed quality management software can also significantly facilitate process improvements by providing easy and reliable access to electronic record preparation and execution across manufacturing sites.
It can also directly contribute to improving the overall quality of products and services and expediting regulatory compliance.
The fundamental requirements of all software systems are the same.
We input raw data through a user interface. The software application analyzes and processes data in various logical programs developed by humans.
The outcome of data processing is to generate meaningful reports through the application that facilitates making essential decisions.
The reports generated by the software used for quality management must be robust, reproducible, trustworthy, and complete. Otherwise, they will adversely affect product quality, and patients will suffer. Hence, regulatory agencies are imposing stricter requirements to develop GMP compliant software.
The software should have functionalities such as electronic signature and record management, a list of records by registered date and time, and strict access control between common users, reviewers, approvers, and administrators.
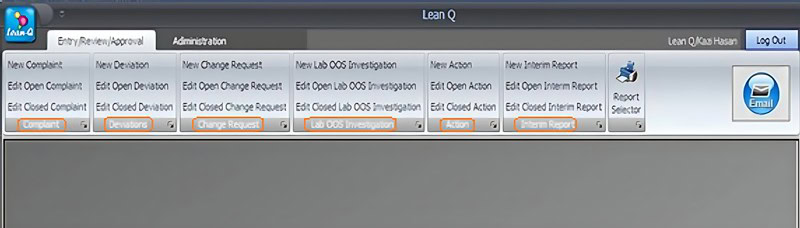
You can design the software application using a modular approach. Each module should be dedicated to one specific area of investigation, such as complaint handling or deviation management.
Users can access those modules independently from one integrated user interface supported by a remotely hosted physical or cloud database.
The database should be able to contain an extensive library of information (e.g., users, raw materials, components, intermediates, finished products, departments, facilities, equipment, documents, batch numbers, test methods, specifications, processing lines, suppliers, and many more items as required).
Although the individual software modules will be very specific to their own investigation processes, information from the database library will be used by all modules.
As I explore this topic further, I will explain more about preparing specifications, user requirements, development, and qualifications. I will also provide some examples to help you understand how to manage each module in compliance with cGMP (Current Good Manufacturing Practice) and other regulatory expectations.
The software should also support the principles of Good Documentation Practice (GDP), have a built-in audit trail (record history) system, continuous database backups, a multipurpose useful report generation system, including trend analysis, and more.
The application should be simple to operate and efficiently process large numbers of data in the background. Automating report generation, including quality trends, activity audit trails, and electronic record management, should provide the users with a GMP compliant environment.
The software should significantly reduce the cost of maintaining quality, reduce manual data processing time, boost users’ responsibility and accountability for maintaining GMP, and help transform conventional hard copies into electronic record management.
Regulatory basis for quality management software
Over the last few decades, regulatory agencies from several countries have made significant efforts to guide their life science industries in developing and maintaining validated electronic systems that can be used reliably for electronic record generation and management.
One of the most popular guidelines in this area is the United States Food and Drug Administration (USFDA), part 11 of Title 21 of the Code of Federal Regulations, “Electronic Records & Electronic Signatures” (hereafter to be called 21 CFR Part 11).
In this guidance document, the FDA has made specific recommendations for manufacturing businesses producing therapeutic products on developing, modifying, maintaining, archiving, retrieving, and transmitting electronic records.
The requirements outlined in 21 CFR Part 11 also include certain provisions of the Current Good Manufacturing Practice regulations (21 CFR Part 211), the Quality System regulation (21 CFR Part 820), and the Good Laboratory Practice for Non-clinical Laboratory Studies regulations (21 CFR Part 58).
However, I will focus on the requirements for electronic records and signatures described in 21 CFR part 11 documentation. These requirements are important to follow while developing GMP compliant software applications.
Summary of 21 CFR Part 11 requirements
The rules and recommendations for maintaining electronic records governed by 21 CFR Part 11 can be summarized and interpreted as follows. Businesses should take adequate steps to:
– Limiting the electronic system access to authorized individuals.
– Operational system checks of the computerized systems are used.
– Perform authority checks for using those computerized systems.
– Use of device check.
– Determine that persons who develop, maintain, or use electronic systems have the education, training, and experience to perform their assigned tasks.
– Establish and adhere to written policies that hold individuals accountable for actions initiated under their electronic signatures.
– Appropriate controls over systems documentation.
– Controls for open systems corresponding to controls for closed systems.
The 21 CFR Part 11 also tells us that:
i. Records that are required to be maintained can be in electronic format in place of paper format or in electronic format in addition to paper format and must be relied on to perform regulated activities.
ii. Businesses should exercise caution and take a risk-based approach to validating their electronic systems to ensure the accuracy, reliability, integrity, availability, and authenticity of the required records and signatures.
iii. Persons working for the business must comply with the requirements related to the maintenance of electronic documentation with an audit trail, which should demonstrate the person’s name, position, time/date stamping of an electronic signature, and the sequence of events are maintained to ensure changes to a record do not obscure previous entries. The availability of an audit trail ensures the trustworthiness and reliability of an electronic record.
iv. Businesses should be able to produce copies of electronic records to be held in common portable formats when records are kept in those formats, and businesses should be able to provide an investigator reasonable and useful access to the records during an inspection.
v. Businesses must be able to adequately protect their electronic records and retrieve them accurately and readily throughout the record retention period.
250 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents are included each month. All written and updated by GMP experts. Check out sample previews. Access to exclusive content for an affordable fee.
What are the quality assurance investigations?
Pharmaceutical businesses routinely conduct multiple quality concern investigations, such as deviation investigations, out-of-specification result investigations, product complaint handling, and CAPA investigations.
A typical quality concern investigation report should involve at least the following:
– Brief description of the subject to which the problem relates.
– Description of the incident/problem/deviation/complaint.
– Documentation of probable causes of the incident.
– Documentation of investigation findings relative to each probable cause.
– Documentation of evaluation of each probable cause.
– Elaboration on actual or suspect cause(s).
– Documentation of the investigator’s logic; citation of the data and information being used.
– Evaluation and documentation of whether other batches are potentially involved.
– Documentation of conclusions and rationales.
– Outline of corrective and preventive actions; short-term & long-term.
– Outline of impact on status of disposition of batches, etc.
Typical investigation process steps
A typical quality investigation process should involve the following sequential process steps, which should be managed by the software application.

250 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents are included each month. All written and updated by GMP experts. Check out sample previews. Access to exclusive content for an affordable fee.
What capabilities are expected from GMP compliant software?
As mentioned before, well-designed quality management software should reliably complete quality investigations in electronic format, eliminate errors and duplications, and maintain regulatory requirements.
The effectiveness of quality management software depends on comprehensive user specifications and the capacity to build a system from the ground up based on these requirements.
The specification should be designed to comply with all regulatory requirements and automate tasks that would otherwise be performed manually.
The software specification should include the following items but not limited to:
i. The GMP software has built-in functionality to securely log in only by authorized users. A strong identification and password algorithm is needed to log into the program.
ii. It should have multiple layers of permission levels among the users, such as system Administrator, entry-level, Reviewer, and Approver. This ensures that authorized persons can only perform specific tasks according to their positions and responsibilities.
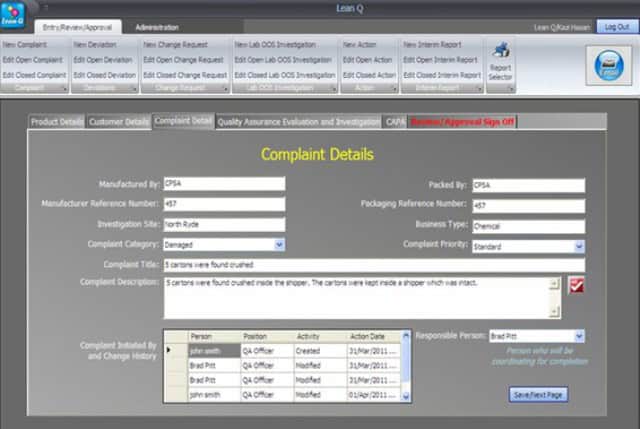
iii. The software investigation forms must document each step of an investigation according to its sequence of events: for example, record initiation > investigation > record review > record approval > and record closure.
iv. The software should be designed to reflect an investigation’s real-time workflow sequence, not allowing a task to be performed earlier and breaking the logical sequence.
v. All new entries and amendments to old entries during the investigation process must be saved after an authorized person executes an electronic signature. All electronic signatures accompany the person’s name, position title, date, time, and activity comment. The signature and entry comments are displayed on the live record and can be searched for reports.
vi. The software should not allow the users to leave or skip a field blank (which has significance on the investigation workflow) to comply as a GMP document.
vii. It needs to be designed with numerous validation rules that guide the users to complete a task sequentially and in accordance with GMP. An automated audit system checks the entries on the form and prompts the user if any of those validation rules are violated during an investigation. The application should provide recommendations to users where the rules are broken and the necessary steps to correct them.
viii. The software should not allow users to delete a comment or step from an approved and closed record unless permitted by senior management or administrators.
ix. It should be designed to prevent the administrator from deleting a user from the system who no longer works for the organization. This ensures the record retention provision described in the FDA regulation 21 CFR Part 11 is met.
x. The database should be hosted on secured servers with restricted access only by experienced developers. As a contingency plan for disaster recovery, the database should have the option to be backed up on its server and possibly on a remote server.
xi. The software should be able to generate records, analyze reports based on true data, and generate copies of the reports that can be stored in portable electronic devices.
Design and development of software user interface
i. Application downloads and installation
There are many ways to deliver a software application. You should provide an easy option for users to download and install the application on their laptops or desktops and make it compatible with the most popular operating systems and browsers.
The application should run on a network environment through a client-server setup with a centrally installed database and application.
The installation process should be automatic and in Wizard format. It will start when the end user downloads and installs the files from an installation shield.
Setting up an application Super Administrator should be part of the installation process.
ii. Database security and backup
The database should be built securely, and users should not have access to the central database. The end user should not be able to add/update or delete any part of the database.
The user should contact the Administrator or IT team through an online help ticket for any application-related issue.
The database should have the option to be backed up at a frequency set by the developer. This will serve the purpose in case a server or database collapses.
iii. Help and support
A help file or training support should be accessible from the application interface. It should detail the exact workflow of each module and section of the application to help the user operate the program with minimum difficulty.
iv. User login
The user must log in to access the application’s user interface. They must enter their authorized ID and password to access the software.
v. Login failure
For a failed attempt to access the application, an error message will appear as follows:


250 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents are included each month. All written and updated by GMP experts. Check out sample previews. Access to exclusive content for an affordable fee.
vi User authentication
You can establish a process of user authentication by limiting the number of failed login attempts. For example, over five failed login attempts should lock the system from an unauthorized user or inactivate access for an authorized user.
Only the Admin should be able to reactivate the user. A message such as “Incorrect attempt exceeded limit” should appear for successive login failures. “Your account has been locked. Please contact Admin”
vii. Auto logout
If the users stay inactive for more than 15 minutes, they will be automatically logged out of the application and must log back in.
viii. User password management
The user should be able to change the assigned password from their profile anytime. For additional security, you can mandate that users change their passwords at a certain frequency through a “Change Password” link. Fifteen days before the password expires, the user will be prompted automatically to change the password.
You can establish a password length between 6 and 12 characters in alpha-numeric format. You can also mandate that the user not repeat six consecutive old passwords while creating a new password.
ix. Electronic signatures
A signature dialogue box will appear when a user performs an action on the open record and decides to save new information. In the dialogue box, the users have to write a comment on the action they have performed, followed by entering their ID and password. Filling out the comment section should be mandated. Users will be notified that their signatures are legally binding.
Only authorized users should be able to sign in to complete an activity.
A new electronic signature comprises the user’s name, position, and date and time of the signature. The generated signature will appear on the record.
If multiple users amend different sections of the electronic form, as per their permission level, the relevant signatures will populate on top of the previous signatures and appear on the form. The log will be shown as signature history on the electronic form and can be printed when required.


Activities performed inside the investigation modules
Users can perform three primary activities inside each investigation module.
i. Raise a new record:
After successful login, any authorized user should be able to raise a new record (complaint, deviation, change request, etc.) by opening a blank electronic form relevant module.
In general, each investigation electronic form can be processed in five steps that are arranged sequentially:
– Record initiation.
– Investigation process flow.
– Review of investigation.
– Approval of investigation, and
– Closure of investigation.
A new record number will be generated as the new form is initiated, filled out with analytical information, and saved using an electronic signature.
The number format should be specified during software development. It is important to choose a format that resembles the type of investigation conducted and should not instigate confusion among different types of investigations (e.g., COMP 001 or DEV 001).
At the end of the investigation, authorized persons should be able to edit, review, approve, and close the record according to their permission levels.
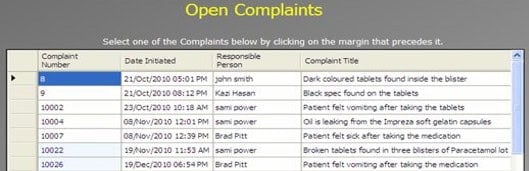
ii. Edit an open record:
In this section, the user can select an open investigation (record) that has yet to be completed. The user can add, amend, and save new information using the electronic signature principle within the validation rules.
Note: Users cannot edit closed investigations. Only the Admin can re-open a closed investigation (e.g., complaint, deviation, change request, or CAPA) with a specific rationale.
Once a closed investigation is reactivated, the user can edit and save as per electronic signature requirements. The investigation information must be re-reviewed, approved, and closed again.
iii. Edit a closed record
Most users should not be allowed to edit a record already approved and closed by authorized persons. This option will be exclusively reserved for the System Administrator, which means only an Administrator should open a closed record and assign a responsible user to edit it if necessary.
When users open and edit a closed record, it must follow the same process of reviewing, approving, and closing according to their permission levels. The entire sequence of activities must be logged into an audit trail, which cannot be amended.
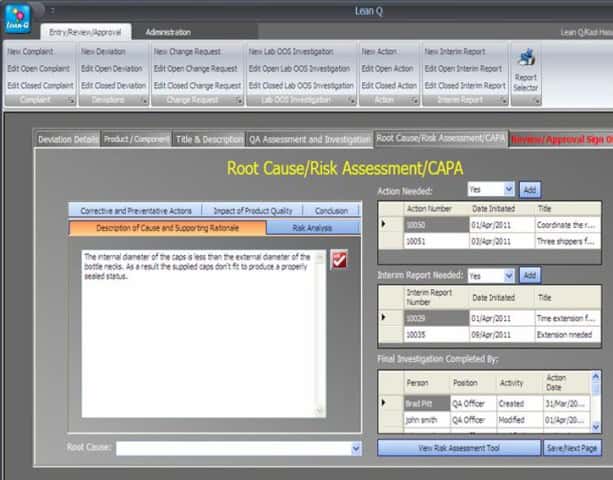
iv. Complete a root cause analysis
The user should be able to conduct a root cause investigation and risk assessment and take corrective and preventive actions inside the investigation module. This automation can greatly improve the department’s productivity.
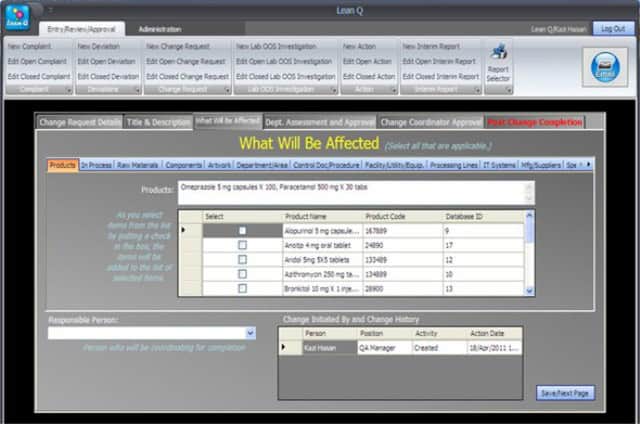
v. Conduct change control assessments
The user should be able to conduct a change control assessment within the Change Management module and suggest what system, process, facility, equipment, or documentation will be impacted by the change.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
250 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents are included each month. All written and updated by GMP experts. Check out sample previews. Access to exclusive content for an affordable fee.
Features inside the software administration panel
The software administration panel should only be accessible by an authorized Administrator. Super Administrator can create multiple administrators as required by the business organization.
The Administrator/s can create as many users as the business organization requires.
Creating a new user and administrator
Only a Super Administrator or an Administrator can create a new user with different permission levels for specific modules. A new user can perform tasks according to the permission level, i.e., create a new record, enter data, edit existing information, review, approve, and close the form.
However, a user should be permitted to edit, remove, or change any part of the closed record. Only the Administrator has the authority to open and edit a closed record.
i. Create a new administrator
Creating a new Administrator is the same as creating a new user from the Administration panel. A checkbox for Administrator is built to determine which users have Administration authority.
ii. Setting up user groups and permission levels
An Administrator has the privilege to create, activate, and deactivate users. However, an Administrator cannot permanently delete a user from the system.
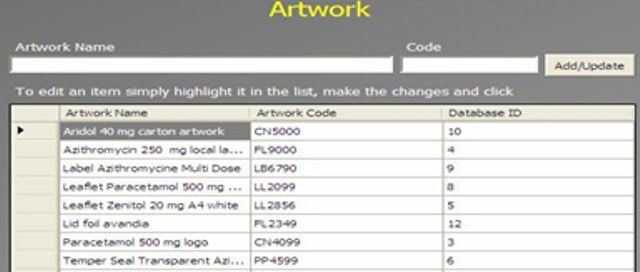
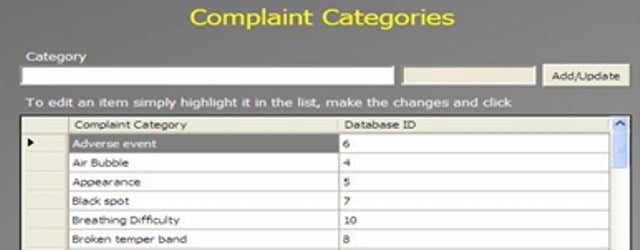
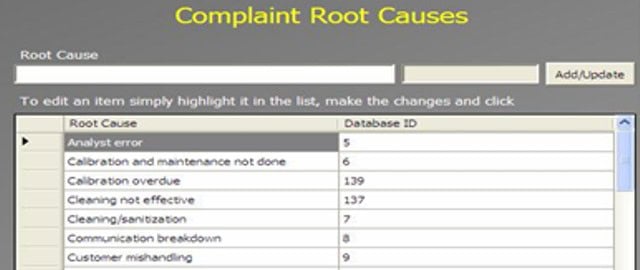
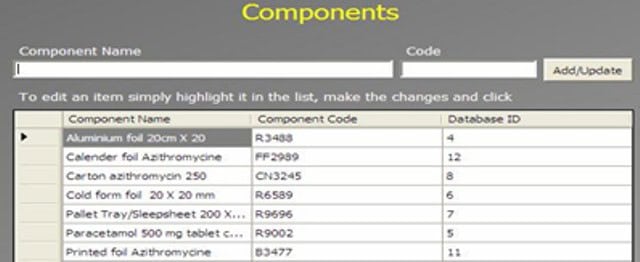
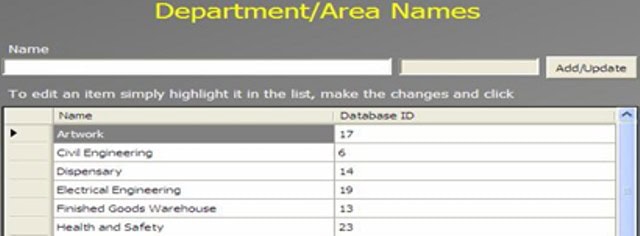
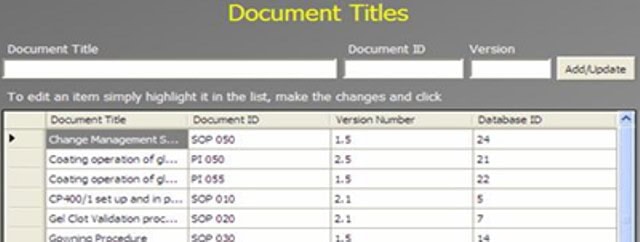
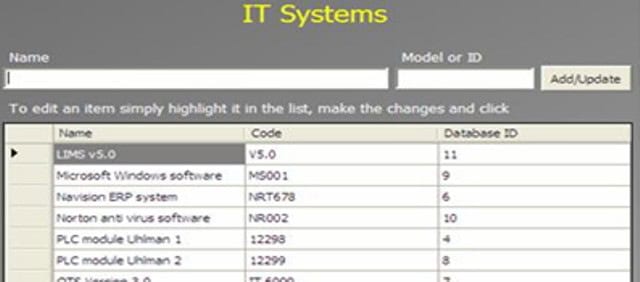
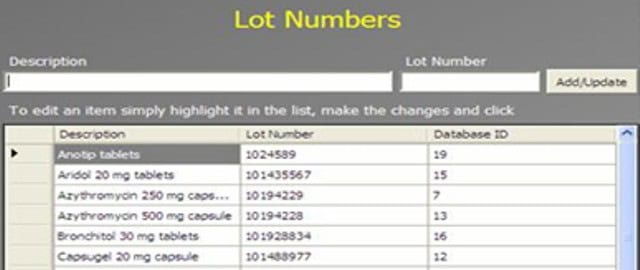
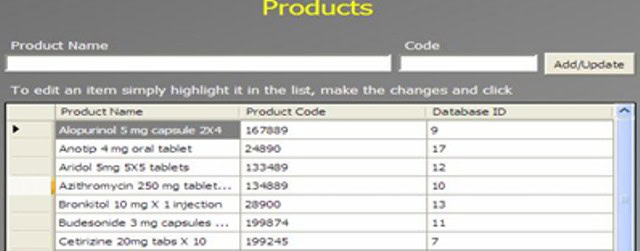
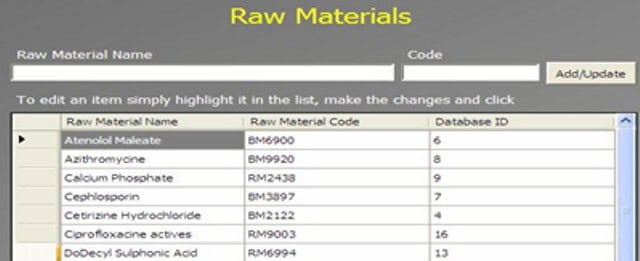
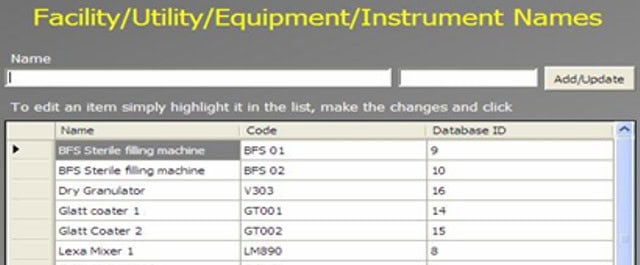
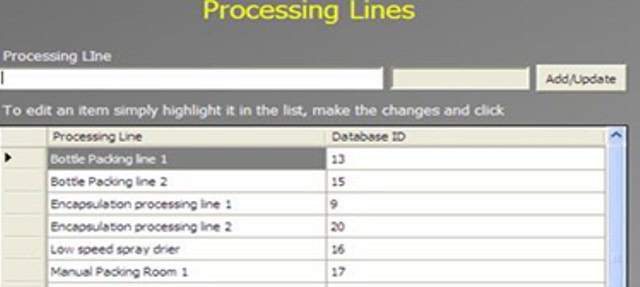
An Administrator can add a new complaint category, root cause, product, raw materials, component, and artwork information. The admin can also add a deviation category, a facility, utility, equipment name, processing lines, a department or area name, document titles, document ID with version number, an IT system name, etc.
Only an Administrator can open a closed record from any of the modules and edit it. The administrator can also permanently cancel a record from the system if necessary.


iii. Entry-level user group:
The Entry-Level user group has permission to generate new records by entering and selecting new data from a blank form. However, entry-level users cannot review, approve, or close a form.
They also do not have permission to see the ‘Reviewed by,’ ‘Approved by,’ and ‘Closed By’ sections of the record as these buttons are deliberately kept inactive for this group.
The “Review/Approval” sign-off should be inactive for Entry-level permission. An entry-level user cannot see an investigation record’s last section/tab.

iv. Reviewer user group:
The reviewer group has permission to generate new records by entering and selecting new data from a blank form and can review the work performed by the Entry-Level users. However, the reviewer user can not approve or close a form.
These users should not have permission to see the ‘Approved by’ and ‘Closed By’ sections of the record, as these buttons are deliberately kept inactive for this group.
v. Approver and closure user group:
The approver group has permission to generate new records by entering and selecting new data from a blank form. They can also review the work the entry-level and reviewer users perform, approve it, and close a form.
The “Review/Approval” sign-off section should be active at the Reviewer or Approver permission level. A Reviewer or Approver user can see the last section/tab of the record.
Only a reviewer and approver should be able to sign off on a final investigation.

vi. Editing a user
Design the software specification such that an existing user profile can only be edited by an Administrator.
The software administrator should be able to select a list of active and inactive users’ names and statuses from the user interface’s Edit User button option.
When a name is selected, the user’s profile page opens, from where the user’s profile can be changed or edited.
This includes changing the user’s first name, last name, position titles, email address, user password, permission levels to different modules, setting up as an Administrator, etc.
When a user’s profile is edited, the user’s activities and permission levels are changed accordingly.
Creating selectable items in the administration panel
In GMP compliant software, users can select some items while conducting an electronic investigation. Let’s call these items and categories libraries.
Only an administrator should be able to create a new library and include or archive items from the libraries through the Administration panel.
Each library type represents a unique function typical to a manufacturing organization and can be impacted during the investigation stages.
The electronic forms of each module contain a drop-down list with relevant items. While working on an investigation, users can select the items necessary for their investigation.
The administrator can add and update any library item at any time but cannot delete an item.
250 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents are included each month. All written and updated by GMP experts. Check out sample previews. Access to exclusive content for an affordable fee.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Designing features for investigation modules
In GMP software, users can access different investigation modules to raise a new investigation, conduct root cause analysis, edit the investigation with new information, and review and complete those investigations.
When the new investigation is raised, a new form opens with several investigational steps to complete. The software must comply with specific rules and parameters while investigating electronically, and these features are carefully planned when the software is designed and developed.
Primary features of investigation records:
– Investigation numbers should be automatically generated in the format of your choice (e.g., COMP 001 or DEV 001).
– A red asterisk (*) indicates a mandatory field on the electronic form. If the fields are not selected or entered before saving is attempted, appropriate error messages will appear as reminders and guide the user to complete the field.
– A blue asterisk (*) denotes that it is optional for the entry-level user but mandatory for the reviewer and approver. An appropriate error message will appear before signing off.
– An investigation save button will save all data in the fields. The user log-in name will be shown automatically. When saving an investigation, a window should pop up and ask for a comment, username, and password. Only with the correct username and password, the data will be saved.
– A warning message should appear if the user tries to open another record without saving a task, such as “Unsaved data will be lost.”
– The Send email button will pop up the default email list within the network and allow users to send email notifications directly from the software application, telling another user what job is pending.
– The cancel button will cancel any new entry on the open form after a relevant warning message. The cancel function will work precisely the same way as the SAVE button.
250 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents are included each month. All written and updated by GMP experts. Check out sample previews. Access to exclusive content for an affordable fee.
Electronic record validation rules for GMP Software
To make software compliant with GMP requirements, especially those listed in 21 CFR part 11, you must ensure there are specific validation rules that you must implement in your software to ensure the chronology of the activities is done logically. Some of these are listed below.
– Date investigation initiated: This must be the current login date, like any other signature date. (Permission: All Users).
– Initiated by: This date must be the current login date. (Permission: All Users).
– Date QA received: must be the same or earlier than when the investigation was initiated.
– QA assessment by: Like any other signature date, this must be the current login date. It can be the same, but it must not be earlier than “Date Investigation Initiated.” An Error message will pop up otherwise.
If more people sign in to this section in edit mode, their names, dates, and times will appear in the signature field (Permission: All Users).
– Investigated by: Like any other signature date, this must be the current login date. It can be the same, but it must not be earlier than “Date investigation initiated.” An error message will pop up, and the date will not be saved.
If more people sign in to this section in the edit mode, their names, dates, and times will appear in the signature field. (Permission: All Users).
– Protocol executed by: This applies to decisions made during an Out-of-Specification investigation, such as the Re-Sample/Repeat/Retest Section. The signature field can be left optional. (Permission: All Users).
– CAPA initiated by: Like any other signature date, this must be the current login date. (Permission: All Users)
CAPA completed by: Like any other signature date, this must be the current login date. It can be the same date but not earlier than the “CAPA Initiated By” signed off. Otherwise, an Error message will pop up.
– Action completed by: Like any other signature date, this must be the current login date. It can be the same as but not earlier than the “CAPA Initiated By” signed off. Otherwise, An Error message will pop up.
– Investigation reviewed by: This must be the current login date. It can be the same date as “Investigated By,” but it cannot be earlier. An error message will pop up otherwise. (Permission: Reviewer only)
– Investigation approved by: This must be the current login date. This date can be the same but not earlier than the “Investigation Reviewed By” signed off. An Error message will pop up otherwise. (Permission: Only Approver)
– Closed check box: The approver must check this box before closing the form. Otherwise, an error message should pop up, and data won’t be saved.
– Closed by: The investigation closure date cannot be earlier than the “Investigation Approved By” sign-off, and the Checkbox must be ticked. Otherwise, an error message will pop up and data won’t be saved. (Permission: Only Approver).
Generating electronic reports in GMP software
After the investigations are closed successfully, the GMP software application should be able to generate valuable reports for users.
– Preview closed investigation: This will allow the user to select a closed investigation number. Only one investigation report can be opened at a time. This report should have the option to print or send via email in PDF format.
– List of all open investigations: This will generate a separate page listing all open investigations (not closed yet). The report can be printed and emailed to recipients.
– All investigations by time range: This will ask the user to enter a date range first. When the dates are selected, a report combining all investigations for the period selected will be generated. This report can be printed and sent by email to recipients.
– All investigations by product range: This will ask the user to enter a date range and select a product. When selected, a report combining all investigations for specific products for the selected period will be generated. This report can be printed and sent by email to recipients.
– Investigations remain open for over 30 days: Most pharmaceutical companies have a policy of completing a standard investigation within 30 days of initiation. The GMP software will generate such reports, which would be helpful for governance.
– Investigations with action (CAPA): This will ask the user to select a date range. After selection, the software will generate a report listing only those investigations for which CAPA actions were completed. The report can be printed and emailed to recipients.
– Other investigation reports: The software can be designed to generate many useful reports as required by your organization, such as investigations by lot number, processing line, Facility/Utility/Equipment, raw materials, complaint, and deviation categories.
250 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents are included each month. All written and updated by GMP experts. Check out sample previews. Access to exclusive content for an affordable fee.
Conclusion
Good planning and research are required before developing GMP compliant software that can facilitate and automate your quality management system while meeting stringent regulatory requirements.
Transitioning from paper-based systems to electronic solutions will enhance efficiency and compliance with regulatory frameworks like USFDA 21 CFR Part 11.
The software should automate key investigational processes using modular designs and centralized databases, such as complaint handling, deviation management, and CAPA. Core functionalities like electronic signatures, audit trails, and validation checks ensure workflow compliance and accountability.
Effective GMP software development requires a thorough understanding of operational needs and regulatory requirements. Effective design of validation rules, role-based access controls, user activity logs, logical process flows, etc., all but ensure the highest compliance with GMP.
High-quality and accurate report generation is integral to GMP software, as these reports create the baseline for further decision-making.
In the short space of this post, I have tried to explain simple, high-level requirements for designing and developing GMP compliant software. Some requirements can be outdated due to rapidly changing technological landscapes. In real life, the requirements can be very different and complex. However, the fundamentals for quality investigation remain the same. You must consult IT Specialists and internal subject matter experts to plan your software requirements.
References

Author: Kazi Hasan
Kazi is a seasoned pharmaceutical industry professional with over 20 years of experience specializing in production operations, quality management, and process validation.
Kazi has worked with several global pharmaceutical companies to streamline production processes, ensure product quality, and validate operations complying with international regulatory standards and best practices.
Kazi holds several pharmaceutical industry certifications including post-graduate degrees in Engineering Management and Business Administration.