How to use quality risk management in validation testing
- Kazi
- Last modified: March 15, 2025
Quality risk management principles are already effectively utilized in many areas of the pharmaceutical manufacturing business. Quality risk management can be useful in the determination of the scope and extent of commissioning, qualification, and validation activities for systems and production processes in good manufacturing practice.
Instead of going into detail risk management processes, we will try to provide general guidance and examples of where quality risk management can be used in validation disciplines to enable more effective and consistent risk-based decisions. Our scope includes systems and processes that support the manufacture and distribution of active pharmaceutical ingredients and drug products.
Table of Contents
How to apply quality risk management in validation?
Quality risk management has been applied to validation in a number of ways for many years. For example, it can be used proactively to define validation strategy and scope or it can be used reactively to assess the impact of a failure or deviation during validation.
The quality risk management approach correlates what are typical, good validation practices with the ICH Q9 risk management process.
Quality risk management can be used throughout the validation lifecycle, however, some examples of key areas where the application of a risk assessment process brings significant benefits are:
1. Defining priorities for validation activities
2. Defining validation strategy for a process/system, including:
– Identification of critical process parameters (e.g. manufacturing, cleaning, sterilization, depyrogenation)
– Identifying direct impact systems and critical components (e.g. System Validation – System and Component Level Impact Assessments) to determine the scope of qualification.
3. Assessing validation failures/deviations
4. Assessing the impact of changes to a process/system
The use of quality risk management and the methodology to be used should be documented in the relevant validation plan or change management procedure.
When using a risk-based approach in validation, the following provides a correlation of the ICH quality risk management process with the typical activities that may occur during validation.
250 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents are included each month. All written and updated by GMP experts. Check out sample previews. Access to exclusive content for an affordable fee.
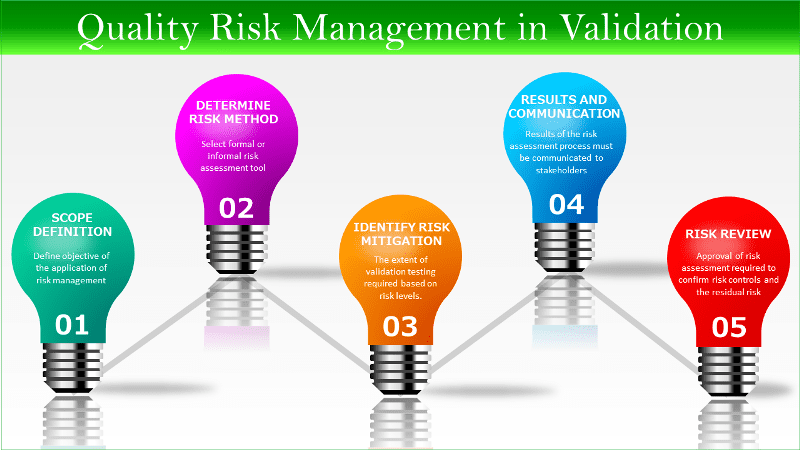
Step 1. Risk management scope definition
The purpose or objective of the application of risk management needs to be clearly understood. Examples of the scope of quality risk management, when applied to validation, include:
– System validation: system boundaries
– Process validation: process definition
– Validation deviation: specific system functionality or process step that has failed and any validation activities that have already been completed.

Step 2. Determine appropriate method of risk assessment
The risk assessment may use a formal (for example, FMEA – Failure Mode Effect Analysis) or informal risk assessment tool. The method should be appropriate to both what is being assessed and the benefits of the assessment itself.
Example: An informal approach, such as a narrative provided by a technical expert that concludes with a risk level, may be appropriate to determine the impact of a validation deviation, as the scope is typically small, and identified risks can be documented as part of the deviation investigation. Identified corrective actions and associated rationale typically provide the risk control/mitigation.
Example: A formal, team-based approach is used for the component-level impact assessment in the GMP site commissioning & qualification process.
A quick method to compare and rank risks, typically involving evaluation of a unique risk event (i.e. deviation / complaint / OOS) by weighting each risk dimension severity, probability and detectability associated with the event.
Examples: Quality concern investigation such as Deviation handling, Product Complaint investigation, Out of Specification investigation.
The tool can also be used for any other quality and compliance issues where a risk event is repetitive and a quality decision is urgent.
A descriptive method to compare and rank risks, typically involving evaluation of multiple diverse quantitative and qualitative factors for each risk, weighting factors and risk scores.
Examples: Manufacturing and Regulatory Change Management; Rework management; Establishing external supplier quality audit frequency.
The tool can also be used for analysing a manufacturing process to identify high risk steps / critical parameters.
Evaluates potential failure modes for processes, and the likely effects on outcomes and/or product performance. Once failure modes are known, risk reduction can be used to eliminate, reduce or control potential failures. Relies upon product or process understanding. Output is a relative ‘risk score” for each failure mode.
Examples: Change of critical instrument Calibration intervals; evaluation of equipment and facilities; quality risk management for supplier; preventive maintenance; process, cleaning and computer validation projects.
Step 3. Identify any risk mitigation requirements
The requirement for validation of both new and existing processes and systems should take into account the risk of the process or system that directly impacts active pharmaceutical ingredients or final product identity, strength, quality, and purity.
This initial, high-level assessment can be used to provide justification as to which processes, systems, or changes will be validated and which do not require validation.
The priority of validation should be documented or referenced in the applicable Validation Plan, validation protocol/s, or change management documentation.
The extent of validation required for a specific process or system should be based on a more detailed risk assessment that evaluates that process, system, or change in terms of its potential to affect product quality. Validation testing may therefore be considered a risk control strategy to demonstrate the robustness of a process or system.
Typically, each process step or system function/component is assessed to determine the level of associated risk. This can then be used to determine the level of testing required to ensure confidence that the system is capable of consistently operating within established limits and tolerances.
A low-risk system component may only require testing during commissioning whereas a high-risk component would need to be verified during qualification.
For example, Validation testing of functions controlling/monitoring critical process parameters or critical quality attributes should be verified during the qualification stages.
A risk-based approach may be used to determine the number of batches required for a process validation study. This includes the use of a bracketing or matrixing approach to process validation to determine which are the “worst case” scenarios or risks. These can be validated to represent the entire range or group.
Risk analysis may be used to create a scientific rationale for environmental control sampling requirements during initial qualification and routine monitoring of sterile and non-sterile manufacturing areas.
For example, during cleaning validation, an evaluation of which residues should be tested should be determined based on risk. The cleaning validation strategy may be based on a “worst case” approach.
The risk assessment enables prioritization of validation activities so that they are focused on the systems, processes, process steps, or system functions/components that pose the greatest risk to product quality. This includes the periodic review and requalification of validated systems and processes.
The risk assessment for a system or process should be prepared based on the planned or current usage, functionality, capability, and capacity of the process or system. It should be maintained through the lifecycle of that process or system.
As mentioned previously, the focus of validation testing on those systems, components, functions, process parameters, and unit operations that have a high impact on product quality is not a new concept and is a fundamental part of many existing validation approaches.
250 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents are included each month. All written and updated by GMP experts. Check out sample previews. Access to exclusive content for an affordable fee.
Typical risk questions to assess during computer system validation
1.0 Category – Product Safety, weight 40%
- Does this system impact or capture data about the quality of the final commercial product or quality of the submission data for the product?
- Does this system contain records that are used in the creation of submissions or in support of manufacturing processes (i.e., training, procedures, calibration, maintenance, investigations, study reports)?
- Is this system used to produce a class A product where company has a significant market share?
- Is this used to distribute the product?
- Is this system used in the creation or verification of product labelling (inserts, outserts, cartons or packaging)?
- Is this system involved in capturing information that would alert the company to the need to take action or support the execution of an action that impacts product quality (e.g. process change, product recall, labelling change, adverse event/safety reporting)?
2.0 Category – System use, weight 30%
- Is this system used to fulfill regulatory requirements?
- Does this system have a history of prior regulatory inspection findings?
- Do the persons who develop, maintain, or use this system have the education, training, and experience to perform their assigned tasks?
3.0 Category – Complexity, weight 15%
- What is the complexity level of the computer system and it's intended use?
- Does this system have interfaces with one or more systems and/or separate and distinct components?
- Does the system implementation require a data migration?
- How was the system developed?
4.0 Category – Technology, weight 15%
- this system a configured application or non configured application?
- What is the implementation scope of this project?
- Is this system/technology new or well‐known for its ability to perform the function?
- Has this system been reliable and stable? (Answer for either case A or B, but not both)? Case A: This system is in place, Or Case B: This is a new system
- Is the vendor who offers this system experienced and stable?
- Is the system networked or stand alone?
- Is a contingency plan in place or being planned for this system should it be nonfunctional?
250 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents are included each month. All written and updated by GMP experts. Check out sample previews. Access to exclusive content for an affordable fee.
Step 4. Document the results and communication
The results of the quality risk management process must be communicated to the relevant stakeholders, including management and those operating the process or system who may be affected by those results.
This requires that each step of the quality risk management process be documented at an appropriate level. The purpose of the output from the risk management process is:
– To share and communicate information about the risks and how they are controlled.
– To obtain the appropriate approval of the decisions taken.
– To demonstrate to stakeholders that there has been a properly conducted systematic approach.
– To provide a record of the risks that enable decisions to be reviewed and the process to be audited.
– To facilitate ongoing monitoring and review and to sustain the process.
The output from the risk assessment must specify a risk owner i.e. a person responsible for ensuring that any actions are implemented and that the risk is managed.
For an informal risk assessment, results and controls can be documented as part of the activity that triggered the assessment, for example:
– Assessment/investigation section of a Deviation form.
– Change Control
Step 5. Approval of the results
i. Risk review
Approval of a completed risk assessment is required to confirm acceptance of the risk evaluation, any identified risk controls, and the residual risk. The final approval of the results of a quality risk assessment must be by the quality authority.
For example:
– An impact assessment for an engineering or information systems project should be approved by the quality representative on the project team;
– A risk assessment conducted as part of an unplanned deviation at the manufacturing site should be approved by the site quality authority.
The risk authority can take either of the following options with a view to the appropriate level of risk mitigation.
ii. Risk Control
Risk control describes the actions taken to deal with the identified quality risks and the acceptance of any residual quality risks. Risk control must address the following questions:
– Is the risk acceptable without further action?
– What can be done to reduce, control or eliminate risks?
– What is the appropriate balance among benefits, risks and resources?
– Are new risks introduced as a result of the identified risks being controlled?
iii. Risk Reduction
Risk Reduction focuses on processes for mitigation or avoidance of quality risk when the risk exceeds an acceptable level. Risk reduction includes:
– Actions taken to mitigate the severity and probability of risk; or
– Processes or methods that improve the ability to detect risk
Implementation of risk reduction measures may introduce new risks into the system or increase the significance of other existing risks.
Therefore, the risk assessment must be repeated to identify and evaluate any possible change in the risk profile.
iv. Risk Acceptance
Risk Acceptance is a decision to accept risk. The risk acceptance decision shall be:
– A decision to accept known, residual risk;
– A decision to accept residual risks, which are partially assessed, based upon limited information; or
– A combination of these circumstances.
The risk assessment (such as a component level impact assessment) for a system or process should also be included as part of the change management for that system or process to ensure that any changes to the intended use or design are assessed appropriately and risk controls are added, removed or modified as appropriate.
Risk assessments that are conducted in response to a specific incident or deviation may be a useful reference but represent a point in time and are not “live” documents, so would not need to be included in change management.
Conclusion
Quality risk management in pharmaceutical manufacturing and validation process can be described as a complete systematic process of identifying validation hazards (risk events), evaluating the potential consequences of those hazards, and controlling the hazards by implementing appropriate mitigation actions with respect to medicinal product quality, purity, efficacy, and traceability.
An optimal quality risk management strategy is designed to reduce risk to an agreed-upon acceptable level. This acceptable level will depend on many parameters and shall be decided on a case-by-case basis and managed through identified mitigation tasks.

Author: Kazi Hasan
Kazi is a seasoned pharmaceutical industry professional with over 20 years of experience specializing in production operations, quality management, and process validation.
Kazi has worked with several global pharmaceutical companies to streamline production processes, ensure product quality, and validate operations complying with international regulatory standards and best practices.
Kazi holds several pharmaceutical industry certifications including post-graduate degrees in Engineering Management and Business Administration.