Change control management – how to implement in 06 steps?
- Kazi
- Last modified: March 13, 2025
“It is not the strongest of the species that survive, nor the most intelligent, but the one most responsive to change”.
– Author unknown – commonly misattributed to Charles Darwin
They say change is the only constant in business. But the business will suffer if the changes are not managed or controlled masterfully.
Imagine your company will install new information management software in the laboratory to replace the old one. The latest software will impact several GMP areas, including the automation of product testing and the release of products to market.
You are asked to raise a change control management protocol. How do you do it?
In the following sections, we will discuss change control management, the step-by-step process to manage change controls at a GMP facility, and some examples especially applicable to the pharmaceutical industry.
Let’s begin….
Table of Contents
What is change control management?
Change control management is a systematic process by which a change to facilities, products, systems, or processes is proposed, assessed by a committee (technical and operational impacts), approved, implemented, reviewed for effectiveness, and communicated to a larger audience.
The change control management process requires a review panel comprised of subject matter experts from cross-functional teams to ensure that all areas of GMP that the change would impact are controlled.
The keywords in any change control management process are compliance with regulatory requirements, impact assessment, change verification, and maintenance documentation.
Why is change control management important in pharmaceuticals?
Change control management is a critical and frequently used concept in the pharmaceutical industry.
For example, pharmaceutical businesses change raw material suppliers for several reasons.
It could be due to supply security, product complaints, improving product stability, material scarcity, supply chain constraints, etc.
Whatever the reason, any change of raw material can directly impact product quality, manufacturing formulae, registration dossier, bill of materials, finished product shelf life, laboratory test method or specification, etc.
Suppose such a change is managed methodically. In that case, all impact assessments are carried out diligently, and risks are identified and mitigated successfully, then the change should not compromise the quality of the finished product.
On the contrary, uncontrolled or poorly managed change will likely bring unpleasant consequences.
Could you take another example?
If you have decided to replace any qualified equipment from the production line, the change can directly impact the validation status, line-specific documentation, employee training, cleaning method, etc.
The ideal way to control such a change is to set up a change management panel to conduct thorough assessments of GMP areas where the change would have direct or indirect impacts.
After the panel approves the change, the equipment can be replaced under the careful watch of a project team.
So that you know, all mitigation actions must be carried out as scheduled.
A minimum of three batches should be run as a trial to ensure no adverse impact on product quality. All documentation must be kept as supporting evidence.
If the equipment replacement (change) is not managed correctly, it will adversely impact the quality and safety of manufactured products. Regulatory requirements will be more challenging to comply with.
Change control management helps maintain product quality and safety by ensuring that any changes made to processes, equipment, or systems are thoroughly evaluated, approved, and implemented in a controlled manner.
This reduces the risk of introducing errors, contamination, or deviations that could compromise product quality, efficacy, or patient safety.
250 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents are included each month. All written and updated by GMP experts. Check out sample previews. Access to exclusive content for an affordable fee.
Difference between change control and change management
Although change management and change control are similar ideas, they have different functions when applied to organizational processes.
The two are different in the following ways:
Change control’s primary goal is to ensure that changes are cautiously assessed, approved, carried out, and documented in a controlled and methodical process.
It entails evaluating the effects of suggested change, identifying potential risks, securing the necessary approvals, implementing the changes, and confirming their efficacy.
On the other hand, change management is a broader concept encompassing a range of activities and strategies aimed at effectively managing and guiding individuals and organizations through periods of change.
Change management aims to plan, communicate, and assist teams and individuals in adapting to change, overcoming resistance, and producing desired results.
It considers the organization’s willingness to change, stakeholder engagement, training, and communication.
Smooth transitions, little interruption, and maximizing the advantages of change initiatives are change management goals.
Change management deals with the human side of change, addressing the psychological and behavioral aspects of individuals and teams affected by the changes, as opposed to change control, which is focused on the procedural and technical components of implementing changes in a controlled manner.
Regulatory Requirements for change management
According to ICH Q10, Change Management is a systematic approach to proposing, evaluating, approving, implementing, and reviewing changes.
– The scope of change management is much broader than change control, typically applied to one change at a time.
– Change management includes overseeing and managing the entire portfolio of changes and the change process, including all the change control components.
– Change management applies across the entire product lifecycle.
A company should have an effective change management system to evaluate, approve, and implement changes.
The change management system should include the following:
– Quality risk management should be utilized to evaluate proposed changes.
– The level of effort and formality of the evaluation should be commensurate with the level of risk.
– Proposed changes should be evaluated against the marketing authorization concerning established products and processes.
– Expert teams, with appropriate expertise and knowledge, should evaluate proposed changes.
– An evaluation of the change should be undertaken after implementation to confirm the change objectives were achieved.
Who is responsible for a change control management process?
All employees within the GMP facility should be responsible for making changes where necessary.
Raising a change does not mean the change will be approved and implemented immediately. All proposed changes must be risk assessed for potential impact on business (GMP) and approved before implementation.
Regardless of the scope of change, you should create a change committee comprising experts in related fields to assess the risks and evaluate the merit and impact of the proposed change.
The change committee must actively participate according to the schedule prepared by the change coordinator so that all potential impacts on products and processes can be assessed.
No stone should be unturned before the change committee approves and implements the change.
Typically, the following stakeholders form part of the change committee. The number and role of the stakeholders may change depending on the nature of the proposed change.
240 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents included each month. All written and updated by GMP experts. Checkout sample previews. Access to exclusive content for an affordable fee.
250 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents are included each month. All written and updated by GMP experts. Check out sample previews. Access to exclusive content for an affordable fee.
The change requester is responsible for:
– Initiating the change by creating a change request.
– Specifying the details of the change, including a description of the proposed change, a justification, and an estimated completion date.
– Obtaining agreement from the department manager of the area affected by the change.
– Ensure adequate supporting data is provided to facilitate approval.
The department managers are responsible for:
– Assessing any change request initiated for their area to ensure that it is value-adding and that system owners have provided all supporting information for assessment.
– Approving each change for implementation.
The change coordinator is responsible for:
– Administering the change management procedure.
– Monitoring the change request that has been raised.
– Advising the plant technical team when changes are ready for review.
– Coordinating the plant technical team meetings.
– Reviewing the deliverables for the completed change request.
– Closing the change request record.
The technical team is responsible for:
– Attending periodic meetings.
– Conducting the impact assessment for each change request.
– Recommending the deliverables and activities required to support the implementation of each change.
– Assigning a project leader and project team.
The Quality Assurance/Compliance Manager is responsible for:
– Approving each change for implementation in the pre-approval stage of the change request.
– Approving completion of each change in the post-approval stage of the change request.
– Assessing employee safety and wellbeing
– Approving each change for implementation in the pre-approval stage of the change request.
The project leader is responsible for:
– Coordinating the project team activities to meet required deliverables as assessed in the impact assessment.
– Extending the estimated completion date of the change request when required using overdue change request notification.
– Compiling the supporting documents to address the deliverables required.
– Notifying the change request coordinator when all deliverables are complete, and change to be closed.
Completing all activities required to successfully document and implement the approved change.
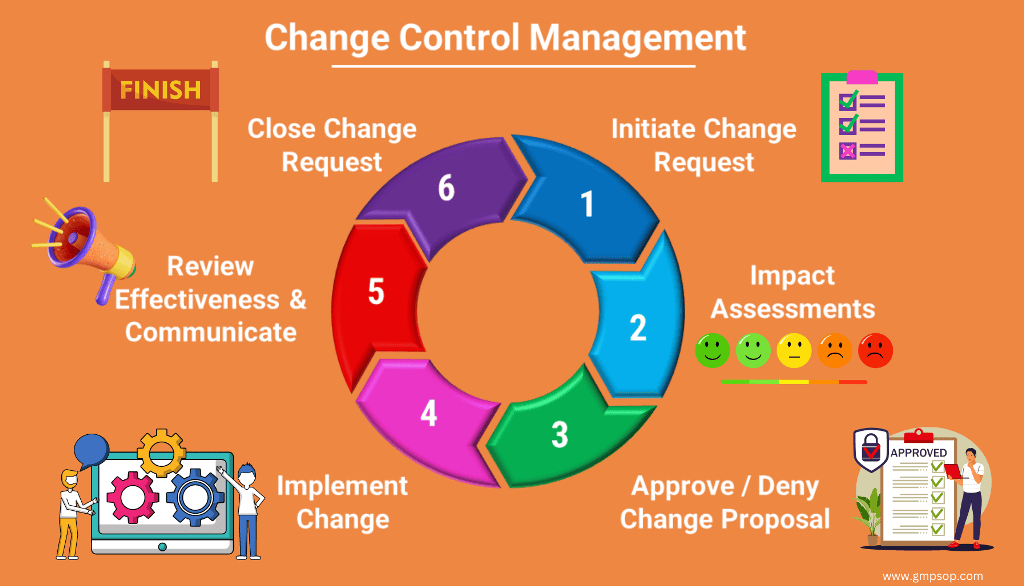
Six steps to follow during the change control process
Change control management is a step-by-step process.
We have identified six steps in a sequence that apply to any change control management life cycle.
If you are responsible for completing a change management process, try the following steps sequentially to complete the change successfully.
Change management process flowchart:

250 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents are included each month. All written and updated by GMP experts. Check out sample previews. Access to exclusive content for an affordable fee.
Step 1: Initiate change request
You can initiate a change by generating a change request. A change request is a draft proposal of change or a group of changes.
A change request can be made using a controlled form or with the use of change management software.
You should include the minimum information on the change request form, such as:
– A description of the proposed change, including scope.
– Where the change will take place.
– The anticipated plan includes a task completion schedule and proposed implementation date (s).
– Potential impact on master documents, i.e., specifications, methods, Formulation, standard operating procedures, etc.
– Estimation of resources required to bring about the change.
– Justification/reason for the change.
– Specify urgency. Enter if the change is either critical/urgent or standard/routine to determine the priority.
i. A critical change will affect quality, regulatory, and safety.
ii. An urgent change will improve cost, customer service, and marketing ability.
iii. A routine change will improve productivity and efficiency.
– Specify if this is a GMP change. Enter Yes or No, depending on the impact on quality management systems and GMP.
– Specify the change type. Enter either major or minor.
i. A minor change is routine without product or process significance. These changes do not require re-validation.
ii. A major change may impact a product’s purity, safety, and efficacy. It may involve changing a critical utility (e.g., HVAC, plant surveillance system, sterile compressed air, etc.) or a process that requires re-validation. Major changes usually require regulatory approval before implementation.
– Change Classification. Please write if the change is permanent or temporary.
– Supplemental documents supporting the change.
– Whether the change requires validation or stability studies.
– Conduct a risk assessment of the change.
– Date of submission of change form.
– Conduct regulatory assessments.
– Implementation information (i.e., date, dependencies, etc.).
– Impact on product supply.
– Multiple/cross-site impacts.
– Assessment of packaging and labeling components.
Additional information may also be needed in the change request form:
– A description of the facility’s status, process, and equipment without the change.
– Available alternatives for the change.
– Approval by an executive group if the change will impact various sites and has worldwide ramifications.
– Any necessary documents, prints, studies, position/concept papers, and data to support the change.
– Subsequent change assessment.
Change management for computer systems may require more implementation information (i.e., change date, first batch impacted, etc.) as part of the change documentation.
If appropriate, you may combine similar changes into a bundle. For example, if multiple systems are impacted, the system changes may be bundled.
Step 2: Conduct impact assessments of the proposed change
You’ll need to conduct a preliminary risk assessment as part of initiating a change request regardless of the criticality of the proposed change.
After submission, the change committee members will conduct separate impact assessments on the areas where they are responsible and knowledgeable.
a. Critical and profound changes (i.e., computer systems):
Please prepare a preliminary technical assessment before presenting it to the change committee.
This would include assessing how the change will impact existing technologies and systems.
How should it be implemented, and what is its estimated development/implementation time?
This technical assessment should include, as appropriate, validation, regulatory and risk assessment, security assessment, ability to reverse the change and recovery strategy, and disaster recovery.
b. Routine and standard change:
Each change committee member must assess the potential impact on GMP, as well as regulatory, environmental, and safety, concerning the proposed change.
Assessors should identify the deliverables their departments require in line with departmental guidelines. They should document the change request form’s final assessment, rationale, and justification.
They should also identify any concerns or special considerations regarding the approval and implementation of the change.
Step 3: Approve, defer, or reject the change request
After formal assessments, the change committee may decide to approve, defer, or reject the request.
The approval process ensures the change is justified, feasible, and aligned with regulatory requirements.
If additional information is required, the change committee should defer the request and seek additional supporting documents from the change coordinator.
Once the requested information is collated, the form should be re-submitted to the committee for further assessment.
The committee can cancel the change request if it is found during assessments that the change would critically impact existing products and processes.
The potential impact on regulatory and GMP will be detrimental and irreversible.
Step 4: Implementation of approved change
You’ll need to start change implementation works after the change committee approves.
After the change is approved, the committee should formulate a project team suitable for actions and generate a realistic timeline for completing all activities.
The project team members should complete each assigned activity according to their departments’ guidelines and procedures.
As the project team completes the tasks, specific details are recorded with supporting documentation.
Once the project is completed, the change request with all records is submitted to quality assurance and regulatory affairs for final approval.
At this stage, the change request will have all the records, assessments, proof of mitigation activities, and supporting documentation to justify the project’s completion.
Step 5: Assess the effectiveness, communicate, and train staff
You should create procedures and protocols to evaluate and assess implemented changes.
I want to let you know that the original change had the intended effect based on the original justification.
The assessment can be performed as part of the implementation process for a minor or evident change.
However, for broader scope and impact changes, the assessment could be done initially and after appropriate data points are available (based on data generation).
User training is critical if there is a major change and multiple systems and processes are impacted.
Prepare your training materials targeting those people impacted explicitly by the changed environment. Keep training material in employee files in line with GMP requirements.
Step 6: Closure of the change
Like initiating a change request, keeping records of change closure is essential.
The change request is closed when,
– The change has been implemented (i.e., installation of new software in the laboratory),
– Documents are updated (i.e., specifications, SOPs, manuals, training, etc.),
– Outstanding actions are completed,
– Follow-up assessments were performed and concluded to be effective.
– Quality, EHS, and Regulatory commitments have been met
– The change is communicated to all stakeholders.
250 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents are included each month. All written and updated by GMP experts. Check out sample previews. Access to exclusive content for an affordable fee.
Change control management examples in pharmaceutical industry
1. Change control management for printed packaging artwork
Printed packaging components are items where GMP critical information for a medicinal batch is printed.
Such as product name, batch number, expiry period, ingredients, composition, concentrations, dosage information, storage condition, age restriction, etc.
Printed packaging components are subject to frequent changes, which may require changes in the registration dossier.
You must carefully coordinate any change in printed packaging components to avoid confusion in patients’ minds.
For the change of printed packaging, consider the following actions:
– Allocation of a packaging artwork number or change of version number.
– Draft artwork and approval by marketing, regulatory, and quality assurance
– Selection of printed packaging suppliers
– Generate or update the Bill of Materials (for packed or finished products)
– Generate or update the specification of the item subject to change for QA release
– Planning assessment of the current stocks
– Timeline of required packaging to arrive (lead time from the point of order to packaging receipt)
– Disposal of superseded artwork in case of the version change (requires a rejection log number & evidence that the disposal has been completed).
– Coordinate with the supplier to obsolete the older artworks, print plates, and knives
– Supplier will update the certificate of analysis or certificate of conformance indicating the updated artwork.
– Keep track of post-change batch numbers and market surveillance
– Update Quality master lists for the impacted item, version, supplier, and GMP agreement (if applicable).
2. Change control management for raw material suppliers and manufacturer
Raw materials used in pharmaceutical manufacturing can be active or excipients.
Active materials have therapeutic actions. The regulatory authority must approve any change of active materials supplier before implementing the change.
Similarly, changes in critical excipient suppliers can lead to changes in the formulation, which may result in out-of-specification results in finished medicine during quality control testing.
If the change in the raw material supplier isn’t controlled, you could likely face unpleasant consequences.
I’ve included the steps for a proposed change to raw material supplier or manufacturer.
– Identify the new alternative supplier based on the performance assessment over price, quality, and reputation/accreditation.
– Perform a quality risk assessment to determine the criticality of the raw material and classify if the material is an active, critical excipient, noncritical excipient, or adjuvant.
– For changes to active and critical excipients, obtain samples taken from three successive batches from the supplier.
– Submit samples for laboratory and microbiology testing to ensure the proposed material meets compendial and in-house physical, chemical, and microbiological criteria established in the specification.
– Submit the change request for initial assessments at least by the Regulatory Affairs, Technical Services, and Quality Assurance teams.
– Technical Services would require 3 x validation trials involving the new source of material followed by complete QC testing to ensure the product meets all quality acceptance criteria outlined in the specification.
– Technical Services and/or Quality Assurance should include the validation batches in the stability testing program for a change of active and critical excipient.
– Regulatory Affairs may require submitting a variation of the registration dossier for supplier and manufacturer change.
– Quality Assurance to assess and approve the supplier and manufacturer through the supplier audit program.
– Quality Assurance to notify the change of the customer as applicable in the GMP agreement.
– Allocate unique item codes if the new raw material is introduced.
– Generate / Update the Bill of Materials for all bulk intermediates (where the material is used in formulation).
– Generate or update the item specification subject to change for QA release.
– Disposal of superseded raw material (requires a rejection log number & evidence that the disposal has been completed).
– Keep track of post-change batch numbers and market surveillance
– Update Quality master lists for the new raw material or impacted raw material (existing).
– Final change control statement of raw material acceptance or rejection based on the trials and stability data.
250 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents are included each month. All written and updated by GMP experts. Check out sample previews. Access to exclusive content for an affordable fee.
3. Change control management for manufacturing processes
Changes to the manufacturing process may involve changes in actives or raw materials, process step substitution or elimination, change in equipment, change of facility or utility, change in testing and release parameters, etc.
If changes to manufacturing processes are poorly managed, it will adversely reflect on the quality and safety of manufactured products. Regulatory expectations will be hard to comply.
I’d like you to follow these steps carefully before implementing a proposed change in the manufacturing process.
– Review the suggested process change with the Regulatory department for impact on registered Products. The final impact must be confirmed by regulations.
– For changes to biological raw materials, the Regulatory Department will assess the change of existing permits to use material and requirements for handling/disposal.
– Changes in active materials need to go through the Regulatory Department so that they can coordinate with local Regulatory authorities.
– Validation will play a big role in accepting these changes and require a validation risk assessment to be completed.
– These changes may be concurrent or prospective and will be determined before implementation.
– Document updates to support change – manufacturing, testing, packaging, filling, specifications, operator instructions, training records, logbooks, etc.
– Update Bill of Materials (where the material is used)
– 3 successful successive trials are required at the pilot/laboratory scale to demonstrate material compatibility in formulation (not applicable for all cases and depends upon the function of the material in formulation).
– Stability requirements for process change (review by stability committee and subsequent recommendation).
– Notifying third-party customers’ approval depends upon the nature of the change.
– Update quality master lists and agreements (if they are in place).
4. Change management during the introduction of new products
As your business grows, you want to introduce new products online to expand your portfolio.
Introducing a new medicinal product or medical device to existing process capability is a major change.
It may require technological changes, additional documentation, new active and excipients, packaging, specifications, and testing regimes.
New products may warrant new facilities or changes to existing facilities, changes in material handling, new compliance requirements, and GMP training for employees, to name a few.
These changes need to be managed by a change committee made up of experts.
The change management project will require several milestones, a work breakdown structure, and a schedule of activities.
Please follow the steps I’ve included below as appropriate to manage the change.
– Which site or contractor site will complete the manufacturing or several manufacturing steps? Does the sponsor approve them, and are they registered for that step with the regulatory agency?
– For changes to biological raw materials, the Regulatory Department will assess the change of existing permits to use material and requirements for handling/disposal.
– You should also consider the market where the target species will use the final product. Regulatory will advise any special requirements specific to the market.
– For re-introducing a previously discontinued product, confirm manufacturing, filling, packaging, testing, and specification documents have not been altered since the last time they were manufactured.
– Regulatory Affairs may require submitting a variation of the product registration dossier.
– Generate a new package of required documentation for new product introduction.
– Publish all required documentation for each manufacturing step, testing, filling, packaging, stability, and release.
– Confirm that all existing equipment has not changed or that new equipment will equivalently process the material.
– All equipment critical to the process must be placed on maintenance and calibration master registers (if brought back in and certified before use in the required range).
– Consider all validation requirements – analytical, cleaning, equipment qualification, process, and utilities. Confirm the impact of change with the validation group.
– Review all raw materials that are going to be used. Where would they be sourced, what quality grade, and how long since the last purchase if unique to the new or re-introduced product?
– Complete three pilot/laboratory scale batches and test against the approved specification.
– Place the pilot/laboratory scale batches on the stability program to demonstrate no un-detectable issue since the last manufacture (especially important if it has been three years or greater since the last manufacture).
– The pilot/laboratory scale batch report must be approved and submitted.
– Confirm all existing suppliers of materials. Complete Approval of Suppliers as appropriate. e.g., supplier audit/questionnaire (need to know if new material is from a new supplier or a new process from an existing supplier).
– Set up of Bill of Materials and allocation of unique item codes (or re-activation if re-introduction)
– The Quality Assurance team should generate or update specifications.
– Place the first batch on the stability program for the re-introduced product.
– Obtain approval from the local Regulatory agency
– Identify all post-change batch numbers.
– Update of Quality master lists.
250 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents are included each month. All written and updated by GMP experts. Check out sample previews. Access to exclusive content for an affordable fee.
5. Change control management of master documents
You can compare the relationship between the master documents and medicinal products with the relationship between a construction master plan and the building itself.
Master documents are files that unambiguously control the quality and reliability of finished products.
What types of documents are considered master documents?
Following is a list of documents that are commonly referred to as master documents:
– Control Method
– Specification (Raw Material/Finished/Packaging)
– Formulation
– Packaging Materials Specifications and Test Method
– Manufacturing Formula
– Manufacturing Instruction
– Bill of Materials
– Stability Specification
– Finished Goods Specification and Test Method
– Raw Material Specification and Test Method
– Standard operating procedures
– Reference of official standards
– Compendia
Any uncontrolled change of the master document can seriously jeopardise the compliance features of your product.
Therefore, you must be extra vigilant and undertake product impact assessment while changing anything in the master document.
New document requests usually result from introducing a new product, new material (ingredient/ components), and new process or procedure or where a need has been identified to a gap with existing documents.
The document change control process follows the usual cycle of proposal, evaluation, approval, implementation, and review of changes.
Revision to existing documents usually results from specification or process changes or corrections. Revision can be initiated and completed using the Master Document Change Control Form.

6. Change control management of computerized systems
A computerized system can be defined by a group of hardware components assembled to perform in conjunction with a set of software programs which, collectively, are designed to perform a specific function or group of functions in a defined environment (and including peripheral devices, personnel, and documentation, e.g., manuals, SOPs).
The computerized system can be any of the items from the following list:
– Application Software
– Operating System Software
– Embedded Software
– Computer Hardware (including microprocessors, PLCs, etc.)
– Networks
– System and Configuration Data
– System Documentation
Changes to implemented computerized systems must always be reviewed, impact and risk assessed, authorized, documented, tested, and approved before the system is returned to operational use.
Conclusion
Change control management is a vital and frequently used strategy in the pharmaceutical industry.
Change control’s primary goal is to ensure that changes are cautiously assessed, approved, implemented, and documented in a controlled and methodical process.
On the other hand, change management is a broader concept encompassing a range of activities and strategies aimed at effectively managing and guiding individuals and organizations through periods of change.
Change management aims to plan, communicate, and assist teams and individuals in adapting to change, overcoming resistance, and producing desired results.
Regardless of the scope of change, you should create a change committee comprising experts in related fields to assess the risks and evaluate the merit and impact of the proposed change.
Every member of the change committee has a very specific role to play.
Change control can be managed in six simple steps: change initiation, impact assessment, approval or rejection, implementation, communication & training, and change closure.
So next time you are assigned to coordinate a change in raw material, packaging component, supplier, manufacturing process, laboratory technology, computerized system, or documentation, you know precisely how to complete the process effectively.

Author: Kazi Hasan
Kazi is a seasoned pharmaceutical industry professional with over 20 years of experience specializing in production operations, quality management, and process validation.
Kazi has worked with several global pharmaceutical companies to streamline production processes, ensure product quality, and validate operations complying with international regulatory standards and best practices.
Kazi holds several pharmaceutical industry certifications including post-graduate degrees in Engineering Management and Business Administration.
we need a simple process of change control system or form or document, since we are only a small laboratory functions