How to Perform GMP Audit in Six Steps
- Published on: Dec 16, 2020
What is the purpose of GMP audits?
GMP audit seeks objective evidence that your quality management system is suitable and continues to be effective by periodically monitoring the performance of an activity or process in accordance with the agreed procedure or work instruction.
You must be continually review and improve your quality management system in order for your company’s products and services to continually meet the customer and regulatory requirements.
GMP audit also seeks evidence that your quality system continues to meet the requirements of the latest cGMP, ISO 9001 and ISO 13485 standards and any commitments made to regulatory bodies.
The results of GMP audit will provide you a powerful mechanism for monitoring and improving the existing quality management system by initiating corrective action when non-conformances or opportunities for improvement to process efficiency are identified.
GMP audits also provide data with which your management can review alongside site mission statement and objectives as well as the effectiveness of the total quality system. GMP audits form part of the company’s key performance indicators.
You should present the GMP audit findings at the management review meetings, where areas for improvement are identified and actioned.
The measurement of such improvements to the quality management system is monitored via regular GMP audits and the existing monitoring mechanisms within the quality system.
How do you perform a GMP audit?
You can perform GMP audits in two ways. On one level, the processes under review are checked for compliance, to ensure that the activities are being performed in accordance with the company’s internal quality procedures and to determine if these procedures in turn meet the requirements of GMP, ISO 9001 and ISO 13485.
On another level, you can take “risk management” approached during GMP audit, where the Auditor examines the effectiveness of the process, procedures and work instructions.
You, as a GMP Auditor must establish that the procedures adequately control the GMP activity and that they have been structured so as to minimise the potential for error.
As soon as you identify an error you must take appropriate mitigation action before the error adversely affecting further process stages.
GMP audits are to be performed in a “process based” manner and you should consider the roles and responsibilities of all process owners and stakeholders.
Auditors shall ensure they seek out evidence to support the adequacy and appropriateness of decisions taken by staff and rationales given.
What is an audit scope and objective?
Prior to conduct a GMP audit you should give adequate consideration to the scope of the audit. This should be limited to manageable parts of the system. Each of such size that the audit can be completed by one or two Auditors in a reasonable period of time whilst also enabling the aspects to be reviewed in sufficient depth.
The scope identifies the boundaries of the processes to be covered during the GMP audit and may include reviews of inter-related processes from other departments if applicable or deemed beneficial. This aspect is viewed as flexible, to allow the Auditor to follow audit trails and adequately investigate potential problem areas when required.
240 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents included each month. All written and updated by GMP experts. Checkout sample previews. Access to exclusive content for an affordable fee.
How do you prepare GMP audit schedule?
The initial frequency and scope of the GMP audits are determined by the Quality Manager and are indicated on the audit schedule. Your company audit schedule should be located on the company network and accessible by the authorised team members only.
GMP audit schedule indicates a date of initial approval, with comments added to indicate reason and dates for any further amendments made. The Quality Manager or authorised delegates are the only people authorised to amend the subject matter, frequency or planned audit as agreed of any item on the schedule.
The GMP audit schedule acts as an initial plan, with the company amending the schedule to take into account activities which may impact on the effectiveness of the quality management system, such as the results of previous audits, significant changes to working practices, the appointment or removal of key personnel etc.
The system audit schedule is based on systematically reviewing the key processes within all of the independently identified departments. Your company should be able to identify which key activities should fall into each of these departments and the activities are then scheduled for audit across the year.
GMP audit schedule also indicate which paragraphs of GMP, ISO 9001 and ISO 13485 should be reviewed during the audit. Audit schedule should include planned reviews of the effectiveness of corrective actions that were implemented previously.
You can use colour code on the GMP schedule to indicate when an audit has been carried out, whether any corrective action requests were raised and also shows when all corrective action have been implemented and closed.
Areas where non-conformances have been recorded during the previous audit may be re-assessed several times during the year to monitor the effectiveness of corrective actions.
Your company Quality Manager shall review the GMP audit schedule for its continuing adequacy. The audit schedule shall be available to all management and department heads via the company network, as formal advance notification of when their area is to be audited.
The availability of the schedule, including records of the results of all GMP audits performed, provides the management team with live data regarding the compliance and efficiency of all the processes within the quality management system.
What documentation and checklist to be used during GMP audit?
During the GMP audit, the Auditor shall consider whether the company procedures continue to comply with the requirements of the standard GMP, ISO9001 and ISO 13485, at the issue that the company is currently certified against.
Your quality manual should list all company procedures for each of the standard GMP elements which need to comply. The Auditor shall ensure that these procedures are being complied with and that all activities undertaken are fully documented within these procedures.
If desired, the Auditor may use a checklist of related questions, phrases or trigger words to guide them through a systematic approach. However, this shall not restrict the Auditor from asking further questions and where a non-conformance is suspected or a process is being performed inefficiently, further detailed investigation must be carried out. The additional details must be attached to the relevant checklist.
Objective evidence for non-conformances found shall also be attached to the checklist for further review where possible. Any checklist raised are to form part of the final audit report. Such check lists need not be distributed to all recipients of the report. It is sufficient to attach them to the report master copy held by the quality manager.
What are the GMP Auditor’s responsibilities?
Your Auditor is responsible to notify in writing to relevant department heads of the forthcoming audit at least one week in advance. Typically, this will be via e-mail communication
This notification shall include the purpose and scope of the GMP audit, date of audit and the applicable procedure/standard reference. The department head should be encouraged to meet with the Auditor prior to the audit date to discuss any particular areas that they wish the Auditor to review.
Should the proposed time for the audit be unsuitable, the department head shall notify the Auditor as soon as possible, in order that a mutually agreed date can be set.
The Auditor is also responsible for generating the Audit Report at the end of the Audit. The Auditor is responsible for preparing the check lists if required.
Personnel performing the GMP audit should not be part of the audit or prepare audit checklists on their own functions. GMP Auditors should be independent of the activity they are going to audit. Departmental personnel can, however perform corrective actions on non-conformances raised, if the activity falls within their normal scope of responsibilities.
What training is required to be a GMP Auditor?
The individuals tasked with performing GMP audits should have a working knowledge of the process and area being audited. Personnel performing the audits should have documented records of internal or external Auditor training supported by certificates.
GMP Auditors must have proven evidence of competency, which indicates they have sufficient knowledge of procedural and regulatory requirements on the GMP areas they are going to audit.
GMP Auditors should understand the basic controls, inputs, outputs and responsibilities and the quality principles involved in different GMP functions.
Your organisation should maintain at least one individual who is trained as a Lead Auditor. The Quality Manager shall determine when Auditor refresher training is required for the trained Auditor and make the appropriate arrangements.
GMP Auditor should also have experience in the human relations aspect of the auditing activity. By correctly utilising this experience it is possible to ensure the most accurate and concise information is obtained from the auditee.
There are several internally reputed GMP audit training providers such as:
What steps should you take during GMP audit process?
240 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents included each month. All written and updated by GMP experts. Checkout sample previews. Access to exclusive content for an affordable fee.
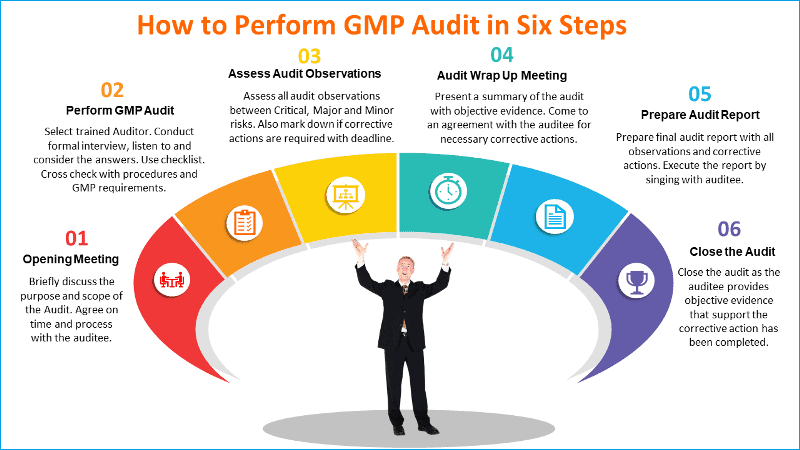
Step 1: Opening Meeting
Prior to commencing the audit an opening meeting should be convened by the Auditor with all heads of the department to be audited, collectively or individually. The purpose of the meeting is to:
– Briefly discuss the purpose and scope of the Audit.
– Confirm the audit program.
– Arrange a tentative time for the closing meeting.
– Explain the audit process and method of reporting.
This meeting does not need to be a formal affair and it is expected that the discussion with the auditees would rather be a short conversation covering the above points.
Step 2: Performing the actual GMP audit
It shall be standard practice when conducting audits to determine if any non-conformances or efficiency improvement opportunities were raised during the last audit on the activity in question.
You, as a GMP Auditor should check any corrective actions applied after previous audit have been effective in preventing re-occurrences.
When performing GMP audit, the Auditors should their skill on questioning, listening and observing.
Auditor shall ask questions to auditee like a formal interview, listen to and consider the answers.
The Auditor should seek and then evaluate objective evidence to verify that what is being done conforms the explanation provided by the auditee.
Also check that the actions are in compliance with the relevant procedure. The Auditor must also consider if the activity is being carried out in the most efficient way.
You, as a trained GMP Auditor must give consideration the level of risk the company is exposed to. For instance,
– Is more than one individual trained to do this particular task?
– If the task is done incorrectly are measures in place to detect this in a timely manner?
By taking this approach, in addition to regulatory compliance, you are essentially carrying out a real time risk assessment on the process being audited.
By this way additional value is added to process audit which leads to improved business efficiency.
Where a checklist has been generated, this shall be used as the basis of the audit. You must record all your observations for evaluation at a later time and include those in the audit report.
Step 3: Assessing GMP audit observations
At the end of the Audit, the Auditor evaluates their observations and any objective evidence obtained during the process.
You should categorise all observations on risk based model and assign them in order of critical, major or minor observations. You must develop a stringent definition for each of the audit criteria and prepare clear management plan if one is reported by the GMP Auditor.
You can follow the below definitions of each of the observation category as below:
Critical Observation: Deficiencies with company standards, and/or current regulatory expectations that provide immediate and significant risk to product quality, patient safety or data integrity or a combination/repetition of major deficiencies that indicate a critical failure of systems.
Major Observation: Deficiencies with company standards and/or current regulatory expectations that provide a potentially significant risk to product quality, patient safety or data integrity, or could potentially result in significant observations from a regulatory agency or a combination/repetition of “other” deficiencies that indicate a failure of system(s).
Minor Observation: Observations of a less serious or isolated nature that are not deemed Critical or Major, but require correction, or suggestions given on how to improve systems or procedures that may be compliant, but would benefit from improvement (e.g. Good Practice seen elsewhere).
Findings which are not compliant against the GMP regulatory requirements should be documented by the GMP Auditor on a corrective action registry and distributed to the relevant department head at the closing meeting or as soon as possible. A copy shall be retained by the Auditor.
Persons responsible for clearing the corrective action shall ensure the actions are carried out before the completion date. Where this is not possible the department shall request in writing to the Quality Manager to grant an extension to the completion date.
Items which are recorded as “Observations” should be noted for review in the next planned audit of the process, to ensure the observations do not become a non-conformance.
Step 4: Audit exit meeting (wrap up)
On completion of the GMP audit a summary of the Auditor’s findings shall be presented to the department heads.
The corrective actions if any, shall be presented and agreed by the auditee. Once agreed it should be left for department head for actioning within an agreed completion date.
In summary, during the exit meeting the GMP Auditor shall:
– Present an objective overview of the Audit.
– Discuss any adverse findings as recorded on corrective actions registry
– Inform Auditee of issue date of the formal Audit Report. (Normally within 5 days of Audit completion).
– Leave any corrective actions for completion
– Thank the Auditees for co-operation and assistance.
Step 5: Audit report
On completion of the GMP audit, you as the Auditor shall raise a detailed audit report. The purpose of the report is to present a concise record of the aspects audited, results obtained, (both positive and negative) and specific action required to be taken.
Step 6. Closure of audit corrective action requests
Wherever possible, auditee must provide objective evidence that supports the corrective action has been completed.
Upon receipt of a closure report and prior to formal closure of audit, the Auditor shall verify if the corrective and preventive action that have been implemented are effective. A review of the continued effectiveness of the corrective action may also be performed at the beginning of next scheduled audit.
What should be included in a GMP audit report template?
The GMP audit report shall be raised in a standard format that should include the following items at the minimum.
A. Cover sheet
Your GMP audit report should start with a cover sheet that typically contains:
– The report number
– The audit type
– The subject of the audit
– The date of the audit
– The name of the Auditor
– The names of the auditees
– The compliance and efficiency rating allocated after the audit*
* This field also contains comparative data (if applicable) relating to the last time the process was audited.
B. Audit Scope
This section contains the particular area, activity or process being reviewed. This section also contains a reference to the key GMP, ISO 9001/13485 paragraphs and company specific requirements and procedures controlling this subject.
C. Audit Approach
This section details the general approach applied by the Auditor to determine the compliance and efficiency of the subject under review.
D. Definitions
This section explains how the findings of each aspect under review are given a classification.
The following classifications are allocated to each aspect reviewed and documented in the report:
Satisfactory:
The aspect complies fully with the applicable regulatory and procedural requirements and is considered fully efficient.
Observation
– The aspect generally complies with all applicable regulatory and procedural requirements but an isolated incident of non-conformance has been found.
– The efficiency of the aspect could be improved.
Unsatisfactory:
The aspect does not comply with the applicable regulatory and procedural requirements.
Several minor non-conformances have been identified, which trend toward a lack of control in the area.
The aspect is considered inefficient and requires review.
E. Scoring mechanism
In order to determine a measurable indication of the level of compliance and efficiency for the process under review, each aspect audited and documented is allocated a points score dependent on its classification.
The classifications are scored as:
– Satisfactory: 100 points
– Observation: 85 points
– Unsatisfactory: 0 points
The total number of points allocated is divided by the number of aspects reviewed and documented. This gives a representative rating score for the subject under review.
F. Audit findings
A summary of the findings of the audit which shall include, both conforming and Non-conforming aspects and also any observations made.
Conforming findings shall be shown as a means of recording documented evidence that the activity has been addressed satisfactorily.
Each entry shall reference the relevant company quality procedure, GMP and ISO 9001/13485 paragraph as applicable.
Where the findings show a Non-Conformance, the Corrective Action Request serial number shall be shown within the report.
G. Audit Conclusion
A brief conclusion should summarise the overall results of the audit and should contain details on any areas of concern and any relevant comments on corrective and preventative actions. This section should also be used for any additional comments the Auditor may wish to make, for example, a significant improvement or deterioration within a particular area of operation or activity.
H. Copies of Audit corrective action
Any audit findings which resulted in corrective action being raised shall be included in the Audit Report.
These copies included in the report shall show as a minimum the Non-Conformance and if already determined and the proposed corrective and preventive action with a proposed completion date nominated by the individual responsible for the action.
Realistic timeframes must be nominated for all corrective actions, which must align with the amount of work required to address the action required. Where possible, actions should be taken within 30 days of the audit taking place.
The distribution of the Audit Report shall include, as a minimum, Key Management Personnel and other Department Heads related to the area or activity being reviewed.
The auditee is responsible for informing the GMP Auditor when corrective actions are completed. The Auditor is responsible for pursuing any actions not completed within the nominated timeframe.
Hard copies of the audit reports are held under the control of your Quality Manager, with soft copies being held in the Senior Management.
Conclusion
In pharmaceutical manufacturing facility periodic GMP audits are mandated in order to maintain company license. GMP audits are performed to verify if the company’s quality management system and processes are performing in compliance with the regulatory requirement as specified in cGMP, ISO 9001 and ISO 13485 standards.
GMP audit should be carried out in a yearly schedule to verify all important aspects of GMP processes, by the trained GMP Auditors. The audit focus can be risk based or process based. During the audit non-conformance observations may immerge. Corrective and preventative actions should be taken to mitigate such risk of non-compliance at the earliest attempt.
GMP audit provides independent review and assessment to ensure systems, facilities and controls used are effective to claim the manufactured products are safe, pure and effective for human use.

Author: Kazi Hasan
Kazi is a seasoned pharmaceutical industry professional with over 20 years of experience specializing in production operations, quality management, and process validation.
Kazi has worked with several global pharmaceutical companies to streamline production processes, ensure product quality, and validate operations complying with international regulatory standards and best practices.
Kazi holds several pharmaceutical industry certifications including post-graduate degrees in Engineering Management and Business Administration.
Related Posts

Basic understanding of quality assurance in GMP
Importance of acceptance criteria in analytical method transfer
How to develop supplier relationship management strategies in GMP



 in GMP 6")





I think that is among the most vital information for me. And i’m happy
studying your article. But want to commentary on some basic things, The website taste
is wonderful, the articles is truly nice : D. Just right activity,
cheers.