Concept of process validation in pharmaceutical industry
- Published on: Apr 25, 2024
What is process validation in pharmaceutical industry?
According to cGMP, process validation in the pharmaceutical industry can be defined as “Establishing documented evidence which provides a high degree of assurance that a specific process will consistently produce a product meeting its pre-determined specifications and quality attributes.”
Process validation is a critical concept in the pharmaceutical industry. Successful validation activities ensure that processes and products meet the required quality standards.
The validation process involves identifying and documenting the specific requirements established to manufacture the product and then demonstrating that the product consistently meets those requirements.
This includes testing the process parameters relevant to making the product and verifying that the process is free from defects and functions as intended.
Additionally, validation includes continuous monitoring and re-evaluation to ensure that manufactured products comply with specifications and the processes involved in making those products comply with established requirements.
In this article, you will learn about process validation, different validation approaches, critical areas of operations where validation is required, and how to conduct a successful validation study.
By implementing robust validation protocols, pharmaceutical companies can ensure that their products are safe, effective, pure, and of the highest quality.
A scenario where validation is required
Suppose your company’s management has recently decided to install an information management system in the analytical laboratory. Your company manufactures pharmaceutical products.
The system has a built-in automation program to log samples, testing, specifications, results, analysts, and reports.
The system needs to be trained to use numerous formulae based on which the system will process the input and generate final results.
Those results will be used to generate a certificate of analysis, and subsequently, the batch of the product can be released to the market.
To prove that the results generated by the new information management system are accurate, precise, robust, reproducible, and free from bias, you have to undertake computer system validation and system qualification studies.
The validation study will not only prove your results are true, but it will also signal the status of product quality and support compliance with regulatory requirements.
240 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents included each month. All written and updated by GMP experts. Checkout sample previews. Access to exclusive content for an affordable fee.
Statistical data on process validation

i. According to a survey by the FDA, 80% of pharmaceutical manufacturers have been cited for validation-related deviations.
ii. According to a study by the ISPE, 70% of pharmaceutical companies reported experiencing delays in product launches due to validation issues.
iii. The FDA estimates that it takes, on average, 8-12 months to validate a new process or product.
iv. A study by the American Association of Pharmaceutical Scientists (AAPS) found that the average cost of validation for a new product is around $100,000.
v. Research by the Parenteral Drug Association (PDA) shows that up to 80% of validation failures are due to a lack of proper documentation.
vi. In a survey by the International Society for Pharmaceutical Engineering (ISPE), 60% of pharmaceutical companies said they had experienced challenges with computer systems validation (CSV).
vii. Research by the Parenteral Drug Association (PDA) shows that up to 50% of validation failures are due to a lack of proper training.
viii. According to a survey by the ISPE, 40% of pharmaceutical companies said they had experienced challenges with process validation.
What process validation approaches are common in pharmaceutical?
There is no one-size-fits-all approach to process validation.
The optimum validation approach you may choose depends on many factors, such as:
i. The types of products your site is licensed to manufacture.
ii. The chemistry and risk profiles of the therapeutic agents you can process.
iii. The complexity of unit operations, processes, and systems you employ during manufacturing and testing.
iv. You might have generated Empirical data, evidence, and experience throughout the process lifecycle.
v. The age and modernity of your facilities and equipment.
vi. The strength of environmental controls you have implemented surrounding your processes.
vii. How well-trained are your manufacturing staff in good manufacturing practices?
These are some of the highlights you’ll need to consider while selecting the validation approach.
Also, you can not ignore the regulatory requirements and guidelines relevant to your products.
Whichever approach you choose, you must build strong justifications and supporting evidence acceptable to your local regulatory authorities.
To keep it simple, three common process validation approaches are prevalent in pharmaceutical manufacturing.
1. Prospective validation
In this approach, you must establish documented evidence that a piece of equipment/process or system will do what it purports to do, based upon a pre-planned series of scientific tests as defined in the validation plan.
Could you perform a prospective validation before you can use a process, method, or system to put into routine use?
In this validation approach, you must prepare a project plan and a well-designed validation protocol with specific tests, parameters, and acceptance criteria.
Conduct the tests, analyze the data, and present the final results to demonstrate that the processes and systems are acceptable in accuracy, precision, reproducibility, and robustness.
2. Concurrent validation
You may select this approach when an existing process can be shown to be in a “state of control” by applying tests on samples at strategic points throughout and at the end of the process.
All data is collected concurrently with the implementation of the process until sufficient information is available to demonstrate process reproducibility.
Suppose you want to concurrently validate an automatic filling machine that has been operating for some time.
During this validation, you need to list the key process parameters, such as approved containers (e.g., tube, ampule, etc.), fill volume and weight accuracy, sealing integrity, and effectiveness of in-process controls.
You can conduct your tests over several routine batches. Collate and analyze all the data and outcomes to demonstrate that the filling machine performs robustly as intended.
3. Retrospective validation
In this approach, you must establish documented evidence that a process does what it purports to do based on a review and analysis of historical data and process performance.
In retrospective validation, you should methodically collect historical data and records.
I’d like you to please look over the empirical data and documented performance to establish confidence that your process has been producing precise, accurate, specific, and reproducible outcomes.
240 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents included each month. All written and updated by GMP experts. Checkout sample previews. Access to exclusive content for an affordable fee.
Which processes need to be validated?
In general, you must formally validate all process steps, production equipment, cleaning methods, testing methods, computer systems, environment, facilities, and utilities that are directly used to manufacture sterile and non-sterile products.
If you have major packaging equipment and processing facilities, those must be validated. This validation is relatively less comprehensive.
All ancillary systems that do not directly impact product quality should be qualified using technical documentation of the extent of the system and how it operates.
Equipment used in pharmaceutical manufacturing processes can range from basic balances to sophisticated and automated process control and monitoring systems (e.g., PLC, SCADA, CIP, etc.)
Since the processes are so different in complexity, it is not practical for you to use a single set of principles to support the same level of validation program. It would also be incorrect from a scientific standpoint.
Suppose you routinely use multiple dedicated software applications with many functionalities. Some you have bought as commercial off-the-shelf and have minimal impact on production operations.
On the other hand, some are specially customized, installed, and configured, directly impacting your processes and product quality.
In such case, you should conduct a risk-based categorization of all processes and systems and list them between,
– Non-Critical
– Commercial off the shelf (COTS)
– Critical, and
– Bespoke
When determining which equipment or process needs validation, follow the GAMP (Good Automated Manufacturing Practice) categorization standards created by the International Society for Pharmaceutical Engineering (ISPE).
GAMP offers a framework for validating automated systems based on equipment criticality.
GAMP categorizes software and hardware according to their complexity, which helps determine how much validation work is necessary.
Follow the diagram below to decide on the extent of necessary process validation activities based on the product risk and criticality of equipment operation on products.

Typical areas to include in validation study
You should include the following processes, systems, facilities, utilities, and computerized systems in your validation plan.
– Manufacturing processes
– Filling and packaging proceses
– Personnel and material flow
– Cleaning procedures
– Testing equipment
– Raw/purified steam
– Purified water
– Compressed Air
– Air conditioning system
– Vacuum
– Power supply
– Lighting
– Cooling water
– Waste etc.
– Computerized Systems Design
– Information system (i.e. PLC, SCADA, LIMS)
– Laboratory automated equipment,
– Manufacturing automated equipment
– Electronic records etc.
Process validation examples in pharmaceutical
As per the local and international GMP requirements, when you adopt a new manufacturing formula or method of preparation, you must take the necessary steps to show that it is suitable for routine processing.
Process validation refers to the product’s performance under predetermined process conditions, demonstrating that the process consistently yields a product of the required quality.
Process validation ensures that manufacturing can produce a consistent, high-quality product.
For example, if my site is licensed to manufacture, test, and release Paracetamol tablets with a label claim of 500mg + 2%, all of my relevant processes and systems that have direct impact on the product must be validated to produce tablets having strengths strictly between 490mg and 510mg.
If my processes fail to consistently produce Paracetamol tablets within those parameters, the validation of my processes will be questionable.
Typical process validation examples include:
– Validation of processing steps.
1. Process validation for formulated products
The process validation of formulated products (e.g., tablets, capsules, ointments, etc.) starts with a well-defined validation plan and protocol.
The process validation protocol outlines the steps to ensure the process consistently produces a formulated product meeting quality standards.
It begins by identifying critical process steps and parameters (e.g., blending, mixing, granulation, tableting, etc.) conducting validation risk assessment and consideration of potential adverse consequences if they are not met.
Historical data or experimentation helps determine the parameters, establishing the proven acceptable range (PAR) within which they can vary without impacting final product quality.
Validation is conducted under conditions mimicking routine manufacturing, with batch size representative of commercial scale.
Sampling and testing ensure compliance with stringent requirements, focusing on critical process steps.
The process validation protocol includes product description, process details, risk assessment, validation approach, testing, and acceptance criteria.
Homogeneity studies are conducted as needed, with sampling plans ensuring representative samples.
Tests demonstrate physical and chemical homogeneity, with individual samples meeting acceptance criteria.
On-line or in-line monitoring may substitute discrete sampling to demonstrate homogeneity or fulfillment of acceptance criteria.
Why do you need three batches for validation?
Your validation study needs at least three batches to provide a more accurate and unbiased representation of the validation model’s performance.
If you use a single batch during your validation study, it may not provide an accurate picture of the process’s performance, as the specific parameters and environments can influence the results of that batch.
When you use multiple batches, the validation results are more likely to represent the process performance on unseen data.
Additionally, more than one validation set should be used to reduce the chance of overfitting and give a better sense of the process’s performance.
2. What is cleaning validation?
Cleaning is the removal of visible and microscopic contamination by dirt, extraneous matter, or product residues by mechanical or physical means.
The purpose of cleaning validation is to provide documented evidence that the cleaning processes used in a manufacturing facility effectively remove residues from a previous batch, cleaning agents, or microbial contamination below pre-established maximum allowable carryover limits.
The goal of cleaning validation is to ensure that the facility and equipment are clean and free from contamination, which can impact the quality of your products.
Evaluation of the effectiveness of cleaning procedure involves sampling of cleaned and sanitized surfaces.
Then, test those samples to determine whether the level of product residues, cleaning agent residues, and bacterial contamination is below a pre-defined maximum allowable carryover limit before the next batch commences.
The term cleaning validation is to be used to describe the analytical investigation of a cleaning procedure. The cleaning validation protocols should provide a rationale for “worst-case” products.
The protocol should explain how the acceptance criteria, chemical and microbial specifications, limits of detection, and sampling methods were developed.
You can read our article “Equipment Cleaning in Pharmaceutical” to explore cleaning validation further.
3. What is analytical method validation?
Analytical method validation is closely related to process validation but is typically carried out separately from process validation.
Analytical method validation, also called methodology or test method validation, involves demonstrating that the analytical methods used to test the quality of your product are accurate, reliable, and reproducible.
This is critical for ensuring the quality and safety of your products.
When you develop your test methods in-house, the method validation process provides more than just documented evidence of accuracy, robustness, effectiveness, reproducibility, and repeatability.
It also offers the opportunity to fine-tune and optimize your methods, leading to even better results and product quality assurance.
The validation protocols should reference background documentation relating to the rationale for determining limits of detection (LOD) and method sensitivity.
Method validation study includes:
– Identification and evaluation of the critical parameters of the analytical methods,
– Perform method validation studies,
– Document & evaluate the results.
To complete an analytical method validation, you will need to validate all or a combination of the following parameters:
– Precision, repeatability, intermediate precision.
– Method accuracy
– Sensitivity: Limit of detection (LOD)
– Sensitivity: Limit of quantitation (LOQ)
– Selectivity/specificity and system suitability
– Method linearity and range
– Method ruggedness and robustness

4. What is computer systems validation?
Computer system validation (CSV) is related to process validation but is carried out differently.
While process validation focuses on the consistency of manufacturing processes in producing quality products, the CSV focuses on supporting those manufacturing processes.
CSV ensures the automated systems are appropriately designed, installed, and performed according to user requirement specifications.
Validation of computer systems provides documented evidence to assure that they will consistently function according to their pre-determined specifications and quality attributes throughout their lifecycle.
Important aspects of computer system validation are:
– The formal management of design (through a specification process).
– System quality (through systematic review and testing).
– Risk (through identification and assessment of novelty and critical functionality).
– Lifecycle (through sustained change control).
Where equipment is controlled by embedded computer systems, computer validation elements may be performed as part of the equipment installation qualification and operational qualification protocols.
Implementing computer and computerized system validation is a structured process that aligns with the Lifecycle model and the principles of process validation discussed in this article.
This alignment ensures a systematic and thorough validation process.
Qualification documentation, in the case of computerized systems, may cover both the controlled equipment and the controlling system, maintaining consistency and comprehensiveness.
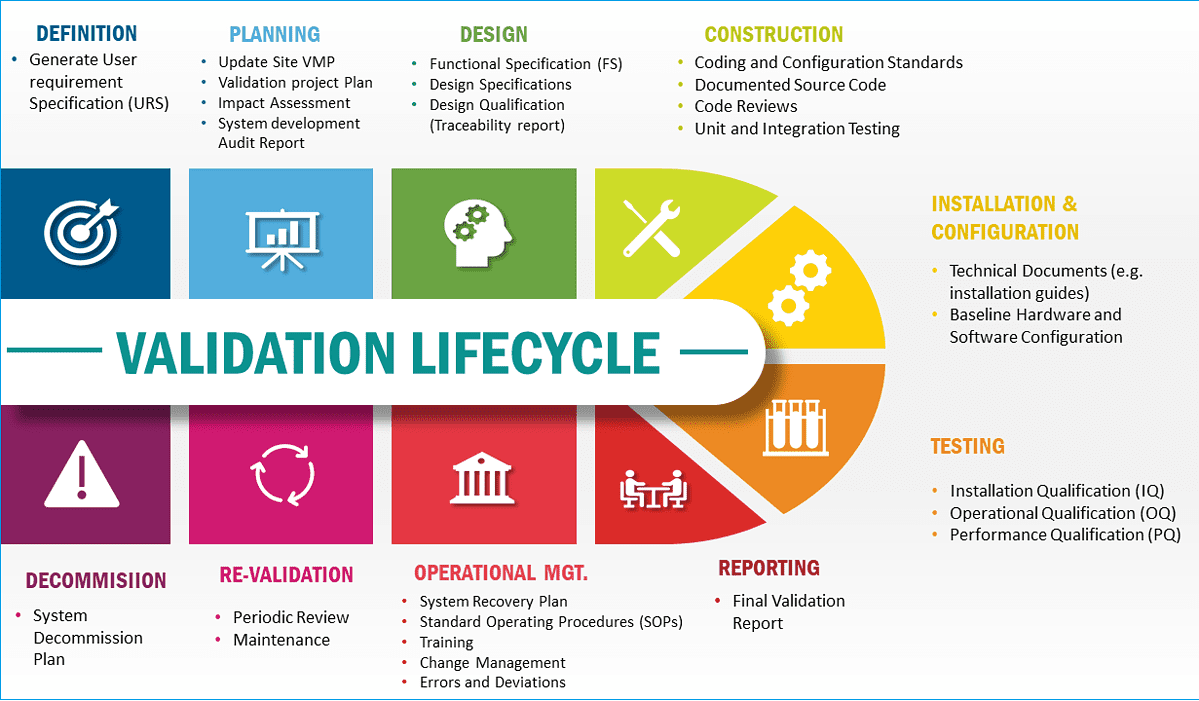
The image below summarizes the validation lifecycle. However, not all phases of the lifecycle detailed below are applicable to all categories of computer systems.

5. What is equipment and facility validation?
Equipment and facility validation, an integral component of process validation, is pivotal in ensuring the quality and safety of our manufacturing processes.
It involves showing that the equipment and facilities you have used in the manufacturing facility are good for their intended use and meet the required quality standards.
The primary objective of equipment and facility validation is to guarantee that the equipment and facilities are functioning optimally, thereby safeguarding the quality of our products.
The extent of equipment and facility validation should be based on risk assessment, such that critical aspects and other system components or functions are verified to be fit for the intended use.
The risk assessment should be based on the impact on product quality and patient safety.
Additional items for consideration include
– The level of standardization,
– Level of complexity,
– Configuration,
– Customisation,
– Intended use, and
– Vendor quality system assessment where applicable.
Equipment and facility validation plans should include periodic reviews of equipment validation status that come in contact with medicinal materials during the production process.
Try our FREE online GMP Skill Booster tests. It’s challanging, it’s refreshing and it’s FREE. Try now!

What steps are taken during equipment qualification?
During the equipment qualification process, you should establish and provide documentary evidence of the following steps:
Step 1: Design Qualification (DQ)
Design qualification intends to produce evidence that the equipment and the processes have been designed in accordance with the requirements of GMP, which include:
– Generation of user requirement specifications
– Verify that the design meets relevant user requirement specifications.
– Supplier assessment /audits
– Challenge of the design by GMP review audits
– Product quality impact assessment
– Specifying validation documentation requirements from equipment suppliers
– Agreement with suppliers on the performance objectives
– Factory Acceptance Testing (FAT), Site Acceptance Testing (SAT) & commissioning procedures
– Defining construction and installation documentation to assist with Installation Qualification (IQ).
Step 2: Installation Qualification (IQ)
Installation qualification intends to produce evidence that the equipment has been built and installed in compliance with their design specifications.
Installation qualification will provide documented evidence that the equipment or system has been developed, supplied, and installed following design drawings, the supplier’s recommendations, and In-house requirements.
Furthermore, installation qualification ensures that a record of the principal features of the equipment or system, as installed, is available and that it is supported by sufficient documentation to enable satisfactory operation, maintenance, and change control to be implemented.
Step 3. Operational Qualification (OQ)
Operational qualification intends to produce evidence that the equipment operates per the design specifications.
The operational qualification will provide you with documented evidence that the equipment operates as intended throughout the specified design, operational, or approved acceptance range of the equipment, as appropriate.
In cases where process steps are tested, a suitable placebo batch will be used to demonstrate equipment functionality.
All new equipment should be fully commissioned before commencing operational qualification to ensure that it is safe to operate.
All mechanical assembly and pre-qualification checks have been completed, the equipment is fully functional, and the documentation is complete.
Step 4. Performance Qualification (PQ)
Performance qualification will provide you with documented evidence that the equipment can consistently achieve and maintain its performance specifications over a prolonged operating period at a defined operating point to produce a product of pre-determined quality.
The performance specification will reference process parameters, in-process, and product specifications.
Performance qualification requires three product batches to meet all acceptance criteria for in-process and product testing.
Performance qualification requires the utility medium to meet all specifications over a prolonged sampling period for utility systems.
The performance qualification documentation should reference standard manufacturing procedures and batch records and describe the methodology of sampling and testing to be used.
240 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents included each month. All written and updated by GMP experts. Checkout sample previews. Access to exclusive content for an affordable fee.
What documentations are critical in process validation?
It’d be great if you could provide appropriate and complete documentation in your effort to process validation.
I’ve included a list of documents you should be able to maintain as supporting evidence of validation activities.
i. Standard operating procedures (SOPs) for equipment, processes, cleaning, and testing methods, which provide instructions for their use and maintenance.
ii. Master validation plan, which outlines the facility’s overall validation strategy and approach.
iii. Validation protocols and reports, validation activities, and results demonstrating that equipment and processes comply with regulatory requirements.
iv. Cleaning validation protocols and reports, which demonstrate that the cleaning procedures used in the facility effectively remove residues of previous products and prevent cross-contamination.
v. Risk management documentation outlines the facility’s risk assessment and risk mitigation strategy.
vi. Equipment and facility qualification protocols with results such as design qualification, installation qualification, operational qualification, and performance qualification.
vii. Manufacturing formulae
viii. Detailed batch documentation
ix. Change control systems
x. Investigational reporting systems
xi. Analytical documentation
xii. Development reports
xiii. validation deviation reports.
It is a regulatory requirement to maintain all validation documentation as evidence that your processes and facilities are operating as intended.
Validation records are used to determine if subsequent process development is necessary.
A revalidation program should be implemented based on a routine equipment revalidation schedule and site change management policy.
What does it take to be a validation engineer?
Certain qualifications are typically required to be a validation engineer in the pharmaceutical industry. These may include:
– A bachelor’s degree in a relevant field such as chemical engineering, pharmaceutical science, or biology.
– Knowledge of the regulations and guidelines set forth by regulatory agencies such as the FDA and EMA for the pharmaceutical industry.
– Experience working in a pharmaceutical manufacturing or development environment.
– Strong problem-solving and analytical skills.
– Strong communication and interpersonal skills, as the role often requires working with cross-functional teams.
– Knowledge of industry-specific software and technologies used for validation, such as computer systems validation (CSV) and laboratory instrument validation.
– Familiarity with Good Manufacturing Practices (GMP) and Good Laboratory Practices (GLP)
– Professional certifications such as Six Sigma, ASQ, ISPE, or GAMP can be beneficial.
Try our FREE online GMP Skill Booster tests. It’s challanging, it’s refreshing and it’s FREE. Try now!
Conclusion
By now, you should realize that process validation is a critical aspect of pharmaceutical manufacturing.
Validation studies offer a remarkable level of assurance, ensuring that your manufactured products are safe, pure, effective, and of high quality for human use.
For robust validation practices, it’s imperative that pharmaceutical companies strictly follow the guidelines set by regulatory agencies like the FDA.
These guidelines encompass requirements for product design, development, testing, and documentation.
You should have a robust validation documentation system to claim your processes are validated and the systems that perform the actions within those processes are claimed to be qualified.
To ensure that validation is carried out effectively, you should develop a comprehensive master validation plan outlining the specific steps to different validation approaches.
This plan should include details on the specific tests that will be performed, the personnel responsible for carrying out the tests, and the timeline for completion.
With the overall knowledge of process validation in the pharmaceutical industry, you can establish a high degree of assurance that your manufacturing processes, activities, or systems will consistently and reliably produce a product that meets specifications and quality characteristics.

Author: Kazi Hasan
Kazi is a seasoned pharmaceutical industry professional with over 20 years of experience specializing in production operations, quality management, and process validation.
Kazi has worked with several global pharmaceutical companies to streamline production processes, ensure product quality, and validate operations complying with international regulatory standards and best practices.
Kazi holds several pharmaceutical industry certifications including post-graduate degrees in Engineering Management and Business Administration.
Related Posts

How to implement good documentation practice in a GMP regulated plant
Control of microorganisms in pharmaceuticals production
A step-by-step guide to successful installation qualification (IQ)


 8")





