Stability testing best practice for Pharmaceutical products
- Kazi
- Last modified: March 13, 2025
Pharmaceutical products must maintain quality, safety, purity and efficacy throughout their specified shelf-life conditions up to their nominated expiration date.
Stability testing ensures that the manufactured products remain safe, pure, and effective throughout their shelf life if they are kept in specified packaging and under environmental conditions.
Stability studies are conducted to:
– Determine the time during which a product meets registered standards when stored under defined conditions (called accelerated studies)
– To show that the product remains within its expiry specifications throughout its proposed shelf life (called real-time studies)
The difference between release and expiry specifications must, therefore, consider the stability testing results.
This article will explore different types of stability studies, designing of stability trial testing program, aspects of stability protocol, different storage conditions, determination of shelf life by analysing stability trial results and common problems that can invalidate a stability testing.
Table of Contents
Key Takeaways
- The main purpose of stability testing is to provide confidence to consumers that the product will remain safe, pure, and effective throughout the shelf life of the pharmaceutical product regardless of the temperature, humidity, or light condition in which it was stored.
- Stability tests are performed inside laboratories to identify the presence of possible degradants or breakdown products impacted by external storage conditions, such as heat, light, or moisture.
- Sometimes, the products are intentionally stored in harsh (accelerated) conditions such as higher temperatures and humidity for shorter periods. The intention is to create a force degradation, followed by stability testing to check how much the product has degraded.
- Real-time (shelf life) studies are performed over the product's shelf life at the recommended storage temperatures. On the other hand, Accelerated studies are performed over a shorter period (3-12 months) at elevated temperature and humidified conditions.
- Stability protocols are required to create a testing regime. It should have the minimum information, such as general product information, specifications and test methodology, results, data analysis and estimated shelf life.
- Bracketing and matrixing help to economise a trial design, such as testing only the lower and upper strengths of formulations, excluding the intermediate combinations of finished products.
- The real-time stability testing time points are generally 0, 3, 6, and 12 months. After that, it would continue through the proposed shelf life. A minimum of three-time points, including the initial and final time points (for example, 0, 3, and 6 months), are recommended for accelerated studies.
- When intermediate testing is required due to significant changes in the product at accelerated conditions, testing at a minimum of four-time points is recommended. For example, in a 12-month study, points would include 0, 6, 9, and 12 months.
- The four climatic zones around the globe dictate pharmaceutical product storage conditions. For example, there are places where the temperature is freezing, subtropical with possible high humidity, hot and dry, and hot and humid conditions. The stability program should be designed to represent each distinct climatic zone.
- Typical stability tests include appearance, physical and chemical attributes, pH, potency (assay), degradation levels, microbiological attributes, and preservative contents.
- Errors that can invalidate the stability results include inadequate specification, incorrect trial design, use of non-stability indicating test methods, inadequate trial monitoring, and incorrect reporting and evaluation of results.
250 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents are included each month. All written and updated by GMP experts. Check out sample previews. Access to exclusive content for an affordable fee.
What is the purpose of stability studies?
The purpose of stability testing has two parts:
i. To provide evidence on how the quality of a drug substance or drug product varies with time under the influence of a variety of environmental factors, such as temperature, humidity, and light; and
ii. To establish a retest period for the drug substance, or a shelf life for the drug product, and recommended storage conditions.
Stability and container closure systems
Stability testing should be conducted on the proposed dosage form that is to be included in the container closure system (including, as appropriate, any secondary packaging and the container label).
Stability and degradation products
Stability studies try to identify the presence of possible degradants in the active ingredient (API) or drug product matrix. Unwanted degradants may be toxic or interfere with the drug’s effectiveness.
i. Origin of degradants in products and APIs
Degradants can come from a variety of sources over time, such as:
– Interactions with excipients in the product matrix
– Interaction with primary packaging through leaching or binding
– Chemical breakdown over time due to heat, pH, light and other factors,
ii. Stability indicating test methods
Chromatography (HPLC) is an important technique used for API and degradant analysis, as it can separate, detect and quantify degradants.
These assays are known as stability-indicating methods. With the appropriate detector sensitivity, column, and mobile phase, stability-indicating methods are very useful for determining degradant concentrations.
Some degradants will have a chromophore, which will make UV detection feasible.
iii. Forced degradation studies – method validation
Forced degradation studies validate the analytical method as stability-indicating in nature. They are necessary for the API testing method and will also have to be conducted for the formulated and packaged product.
iv. Accelerated stability studies
Accelerated (higher temperature) studies are useful for quickly determining degradants and establishing preliminary stability data for the formulation during development.
What is the forced degradation profile during stability testing?
The following chromatography demonstrates a typical forced degradation profile of a pharmaceutical product during high-pressure liquid chromatographic analysis.

Notice that the main active peak (5) elutes at 4.8 minutes and the two main degradant peaks (1 and 2) at 3.8 and 2.9 minutes, respectively.
This indicates that the assay can successfully separate the active from degradants.
The assay then can be called stability indicating in nature and can be used in stability trials.
Peak 2 represents the main degradant, and peak 1 is the secondary degradant.
Analyzing stability data – Kinetic models
A kinetic model for degradation could be used to predict the decomposition rate of active drugs theoretically.
However, kinetic models might not be optimal as they don’t take into account some factors, such as a drug’s matrix or the presence of only a single drug in a reaction.
Dissolution rate, particle formation, and other physical stability issues further complicate.
There is no substitute for determining the shelf life empirically, that is, using real-time data for the formulated product in the final package.

What are the different types of stability studies?
Two types of stability studies can be performed on a product:
Real-time (shelf life) studies are performed:
– Over the entire shelf life of the product
– At temperatures in line with maximum label recommendations for storage
– Using the final primary packaging
– To give a confirmation of stability over the real shell lifetime
Accelerated studies are performed:
– Over a shorter period (3-12 months)
– At elevated temperature/humidify conditions to represent an acceleration of the degradation
– To predict shelf life based on a shorter study period and are used to register the product initially
i. Real-time stability studies
Real-time stability studies are performed on the product at the label condition, such as storage temperature and humidity, while kept in sealed packaging.
The following are the primary requirements for real-time stability testing:
– The formulation of the products on the stability program must be according to the registered details.
– Products on stability programs must be manufactured using all manufacturing steps submitted during registration.
– During stability, the product must be in its registered container and closed. All testing should be performed on this product.
– Initially, at least three batches must be placed on stability, followed by one batch per year.
– Where initial stability was performed on a pilot batch, three full production batches are still required for the same stability trial.
– Drug products of different strengths require at least three batches at minimum and maximum strengths.
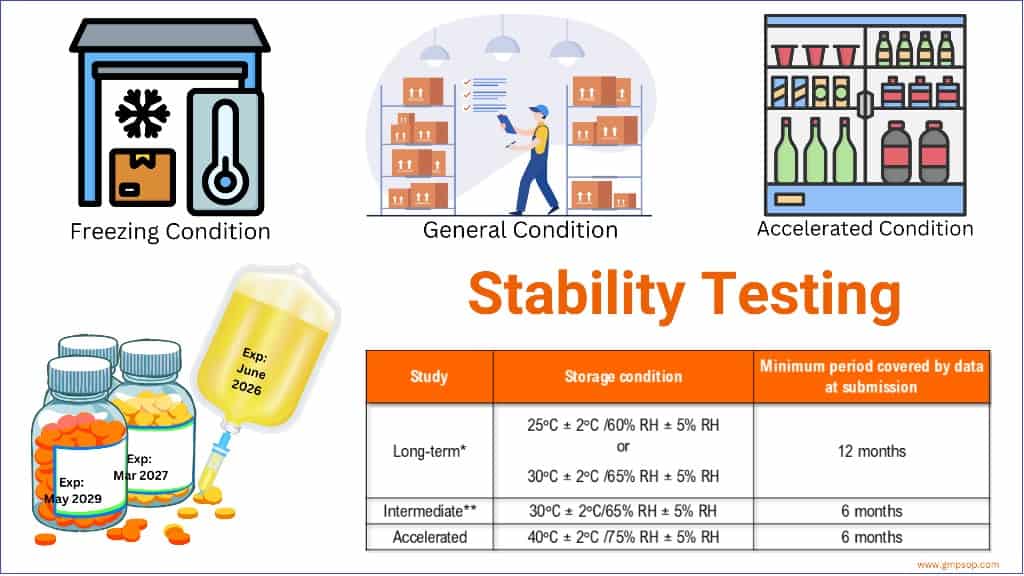
– Temperature (25°C ± 2°C) and Humidity (60% RH +/- 5°C (if warranted) for a minimum of 12 months should be allowed in the studies.
ii. Accelerated condition studies
During stability studies at accelerated conditions, the products were stored in sealed packaging under harsher conditions than specified on the label, such as higher storage temperature and humidity.
Accelerated testing can be performed on development, plot, and validation batches.
The following are the primary requirements for stability testing at accelerated conditions:
– Use a validated stability-indicating method.
– One batch per study is sufficient, as batch-to-batch consistency is not trying to be established.
– Temperature (40°C ± 2°C) at least 10°C above the long-term recommendation) and Humidity (75% RH +/- 5°C if warranted) for a minimum of 06 months should be allowed in the studies.
Additional testing can be performed at an intermediate condition if significant changes are found due to degradation during accelerated testing.
Significant changes can be characterised as:
– 5% potency loss from initial (time zero)
– Degradant approaching or exceeding its specification limit
– Product exceeding its pH limit
– Dissolution exceeding its specification limit
– Deterioration of appearance and physical limits
– Failing to meet specification at 40°C / 75% RH or at 30°C / 75% RH (ICH Guidelines)
250 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents are included each month. All written and updated by GMP experts. Check out sample previews. Access to exclusive content for an affordable fee.
Importance of stability protocols
A stability protocol should be developed and approved by product development, laboratory, quality assurance, and regulatory affairs before any study commences.
The stability protocol should address the following:
– Responsibilities for the trial
– Photostability and stress testing
– Selection of batches and formulations
– Container closure system
– Specifications for the product and tests to be conducted
– Bracketing and matrixing strategy
– Testing schedule (checkpoint matrix)
– Storage conditions and monitoring
– Proposed methods of data analysis and evaluation
– Management of deviations and reporting
A protocol is required for each stability study, with a closeout report once the study is completed.

The protocol would contain the following information:
i. General product information
– Name, source, manufacturing sites, and date of manufacture of drug substance
– Dosage form and strength, including formulation
– Composition, type, source, size of container and closure
ii. Specifications and test methodology
– Physical
– Appearance
– pH
– Organoleptic
– Disintegration
– Chemical
– Chromatographic
– Dissolution
– Degradants
– Microbiological
– Total aerobic count
– Yeast and mould
iii. Study design and plan
– Number of batches, dosage units
– Sampling points, duration, storage conditions
iv. Stability data and information
– Individual data – means, standard deviations
– Tabulated data by storage conditions
v. Data analysis
– Rules for exclusion of data points
– Data, plots, graphics
– Results – statistical analysis, estimated expiry date
250 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents are included each month. All written and updated by GMP experts. Check out sample previews. Access to exclusive content for an affordable fee.
How to design a stability trial testing program?
When designing a stability study, consider the following points:
– Accelerated studies do not necessarily follow kinetics, so extrapolation is often unreliable
– For long-term studies, data can be transformed to get linearity
– Three botches must be tested to assess batch-to-batch variability
– The number of replicates can be tested within a time checkpoint (removes assay-to-assay variation
– Maintain the same number of duplicate estimations at each checkpoint if possible
– Schedule additional assays at the final checkpoint
– Place more samples on trial than required to cover repeat tests
Bracketing and matrixing in a stability program
Bracketing and matrixing for finished dose forms may be used to establish stability trial designs. The ICH have a Guidance for this area, ICH Q1D.
Bracketing allows you to economise by excluding intermediate combinations in a trial design, such as testing only the lower and upper strengths of formulations.
The bracketing design below is based on a product that is available in three strengths and three container sizes. Here, it should be demonstrated that the 15ml and 500ml high-density container sizes truly represent the extremes.
As in a full design, batches for each seeded combination should be tested at each time point.

When does bracketing apply?
Bracketing in stability studies applies in the following situations:
– Where the drug is available in multiple sizes or strengths.
– The range of container/fill sizes and types are the same.
– Range of dosage strengths with very closely related formulations (for example, tablets with different compression weights).
It is not applicable if the formulation is significantly different between strengths; for example, one contains the addition of excipients other than colour or flavouring.
The surface area/volume ratio, dead space/volume ratio, container wall thickness, and closure performance characteristics should be proportional among sizes.
Stability testing frequency and checkpoints
i. Real-time studies
For products, the frequency of testing in the long-term storage condition should typically be:
– Initially
– Every 3 months over the first year
– Every 6 months over the second year
– Annually, after that, it will be through the proposed shelf life.
For real-time product stability, 3 batches are usually tested initially, then one batch per year thereafter.

ii. Accelerated studies
A six-month study is recommended to have a minimum of three time points, including the initial and final time points (for example, 0, 3, and 6 months).

iii. Intermediate Conditions
When intermediate testing is required due to significant changes, a minimum of four-time points are required. For example, in a 12-month study, points would include 0, 6, 9, and 12 months.

250 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents are included each month. All written and updated by GMP experts. Check out sample previews. Access to exclusive content for an affordable fee.
What are the storage conditions during the stability trial?
Storage conditions (with appropriate tolerances) should test thermal stability and, if applicable, sensitivity to moisture or potential solvent loss.
The trial should consider the climatic zones in which the product will be marketed.

Zone 1: Temperature less than 20 0C
Zone 2: Sub-tropical with possible high humidity, averaging 20.5 – 24 0C
Zone 3: Hot and Dry
Zone 4: Hot and humid, averaging more than 24 0C
i. Storage of drug products at general conditions

*It is up to the applicant to decide whether long-term stability studies are performed at 25°C ± 2°C/60% RH ± 5% RH or 30°C ± 2°C/65% RH ± 5% RH.
** If 30°C ± 2°C/65% RH ± 5% RH is the long-term condition, there is no intermediate condition.
ii. Conditions for drug products at cold storage

iii. Conditions for drug products in freezing temperatures

Sensitivity to moisture or potential for solvent loss is not a concern for drug products packaged in impermeable containers that provide a permanent barrier to the passage of moisture or solvent. Thus, according to the general case, stability studies for products stored in impermeable containers can be conducted under any controlled or ambient humidity condition.
iv. Storage conditions for active ingredients

Stability program for active raw materials (API)
For new drug (active) entities, the following information should be available:
– Degradation pathways
– Identity of degradation products
– Suitability of analytical procedures for quantitation of drug substance and degradation products.
Where the degradation pathway of the active is unknown, and therefore the stability can’t be predicted, a forced degradation should be performed, including the effects of:
– Elevated temperature
– Susceptibility to moisture and oxidation
– Light
– pH (if the finished product is a liquid)
250 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents are included each month. All written and updated by GMP experts. Check out sample previews. Access to exclusive content for an affordable fee.
What tests should be included in stability studies?
Stability studies should include testing those attributes of the drug product that are susceptible to change during storage and are likely to influence quality, safety and efficacy. The testing should cover, as appropriate:
– Appearance
– Physical [including weight and water gain or loss)
– Chemical, including pH
– Potency or assay, including degradant levels
– Biological
– Microbiological attributes
– Preservative content (for example, antioxidant or anti-microbial preservative!
– Functionality tests (for example, a dose delivery system)
– Include any in-use conditions, such as the stability of opened packages in multi-dose products.
Expected stability testing for finished pharmaceutical products
Once the stability trial schedule has been designed, the appropriate testing to be performed at each time point needs to be allocated. This will depend on the form and container closure system of the finished dose product.
ICH Guidance Q6A on Specifications directs expected tests for APIs and finished products.
i. Tablets and capsules
– Appearance
– Odour
– Hardness
– Friability
– Moisture content
– Brittleness (hard gelatin capsules)
– weight gain or loss
ii. Liquid formulations
– Appearance, colour
– Odour
– pH
– Clarity (solutions)
– Freedom from visible particulate contamination
– Resuspend ability (suspensions)
– Viscosity
– Phase separation
– Weight gain or loss
– Preservative efficacy
– Closure integrity
iii. Injectable products
– Appearance, colour
– Sterility, pyrogenicity
– Preservative content and efficacy (chemical/biological)
– Particulates
– pH
– Container closure integrity
– Plug appearance and reconstitution time (freeze-dried products)
iv. Ointments and creams
– Appearance and odour
– Viscosity
– Softening range
– Loss of water
– Physical and chemical homogeneity – separation
– Particle size distribution
– Particle formation
– pH
v. Oral powders for reconstitution
– Appearance
– Colour
– Odour
– Moisture
– Reconstitution time
vi. Nasal spray, solutions and suspension
– Appearance
– Colour
– pH
– Clarity (solutions)
– Particulate matter
– Preservatives and antioxidants
– Unit spray medication content, droplet size
– Number of actuation
How do we evaluate the results of the stability study?
You should adopt a systematic approach in evaluating summary stability data and results.
The data and results should include, as appropriate, results from the physical, chemical, biological, and microbiological tests, including particular attributes of the dosage form (for example, dissolution rate for solid oral dosage forms).
The degree of variability of individual batches affects the confidence that a future production batch will remain within specification throughout its shelf life.
i. Physical attributes
– Appearance
– Color
ii. Chemical attributes
– pH
– Assay
– Degradants
– Dissolution
iii. Biological attributes
– Preservative
– Sterility
What to present in the stability results?
While presenting stability results, it is important to include the following:
– Initial (zero time] results
– All individual results, rather than an average
– Fiducial limits per assay for bioassays
– The result as a (%) of the nominal (labelled) content
– Where applicable, individual dose unit variations
– Wherever possible, quantitative results
– Explanations of anomalous or unusual results
– Identify significant changes in the assay method
– Summaries/explanations of the data; rationale for exclusion of data
Review the results after each time point to highlight failures or unusual trends. Investigate any failures using out-of-specification procedures.
Stability indicating methods
Stability-indicating methods can reliably detect changes in chemical composition, such as breakdown products, degradants, excipients, etc., from active pharmaceutical ingredients.
– Don’t assume that pharmacopoeial methods are stability-indicating
– Test methods should be fully described and validated
– If changes are to be made to the assay or other test method during a stability study, it may be difficult to compare results before and after the change
– If two methods are used in the study, compare their accuracy, precision, and specificity
– Changes to dissolution test methodology during stability testing programs are strongly discouraged
– Assays must validate accuracy, precision, linearity, specificity, LOD and LOQ for degradants
– Degradation products present at ≥ 0.5% of the active content should usually be identified
Test methods for stability studies
If an assay is to be used for stability testing, its validation needs to evaluate its stability-indicating nature, particularly with regard to specificity.
The assay should be selective for degradants, not interfered with by matrices, have a defined degradation scheme for all active ingredients, and calculate a mass balance with an aim of >90% contents accounted for.
Stress testing can be performed to determine the degradants. This involves submitting one batch of the active to acid-base hydrolysis, temperature exposure, and exposure to light and oxidising substances.
Microbial stability of products
As well as chemical stability, the microbial stability of products should be assessed by performing the following assessments:
i. Preservative efficacy
– Traditional methods
– Challenge testing or PET test
ii. Chemical stability of the preservative
iii. If a product is multi-dose, simulate consumer use conditions, that is, opening and closing studies.
Microbial challenges only need to be performed at the start, annually, and at the end of the study. However, chemical assays of preservatives should be performed at more regular time points.
Microbial recovery methods should be validated before use in a stability study.
Sterility testing of sterile products is usually performed at the start, 12 months and end of shelf life. Pyrogens/endotoxins should be tested at the beginning and the expiry date.
250 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents are included each month. All written and updated by GMP experts. Check out sample previews. Access to exclusive content for an affordable fee.
Typical problems with stability programs
Sometimes, inadequate data and incorrect interpretation of test results can become problematic in evaluating stability studies. The following is a list of common occurrences that you should avoid when evaluating stability studies.
– Use of expressions such as “Passes Test” instead of figures.
– Deleting out-of-specification points without investigation.
– Failure to provide data from each time checkpoint to assist in the assessment of trends.
– Failure to include either quantitative or semi-quantitative data on degradation products.
– Failure to provide results for individual dosage units.
– Attempting to extrapolate data obtained beyond reasonable limits.
– Failure to comment or conduct further testing when there is a lack of mass balance between forming degradation products and losing the active substance. For example, is the product volatile, or is it absorbed onto the walls of the container?
Stability programs for pharmaceutical products are lengthy and complicated.
A number of things can go wrong, making the program’s validity questionable. Below are some areas where problems can arise.
i. Inadequate specification of products and batches
The following issues can cause the stability trial to be rejected if the product and batches are not clearly defined:
– Failure to specify the formulations used in the trial and to state which batches were identical to the registered formulations.
– Failure to state whether the batches used in the trial were pilot or production batches.
– Failure to describe clearly the packaging used in the trial and whether it is identical to the pack which will be sold.
ii. Inadequate trial design
– Failure to accumulate stability data on more than one batch.
– Failure to accurately define the temperature, lighting and humidity conditions applied during the trial.
– Failure to include information on the physical characteristics of the product during storage.
iii. Inadequate test methods
– Failure to describe test methods, provide validation data on test methods, or even validate the test methods.
– An HPLC procedure is used to detect impurities without validation of the method for that purpose, or the method is only validated for the active ingredient.
– The methods developed for active ingredients are often unsuitable for separating impurities and degradation products. A method must be validated for impurity detection.
iv. Inadequate monitoring of trials
– Failure to conduct studies under the correct humidity conditions.
– Failure to conduct additional testing into obvious changes in the physical characteristics of the product during storage, such as colour changes. Colour changes could indicate the formation of degradation products.
v. Inadequate in reporting and evaluating results
– Use of expressions such as “Passes Test” instead of figures.
– Deleting out-of-specification points without investigation.
– Failure to provide data from each time checkpoint to assist in assessing trends.
– Failure to include either quantitative or semi-quantitative data on degradation products.
– Failure to provide results for individual dosage units.
– Attempting to extrapolate data obtained beyond reasonable limits.
– Failure to comment or conduct further testing when there is a lack of mass balance between forming degradation products and losing the active substance. For example, is the product volatile, or is it absorbed onto the walls of the container?
250 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents are included each month. All written and updated by GMP experts. Check out sample previews. Access to exclusive content for an affordable fee.
How to determine product shelf life from the stability testing?
General rules for evaluating stability data and nominating shelf life include:
i. If no trends are noted after storage for a period of (x) months at an elevated temperature (at least 10C above the maximum storage temperature proposed for the product), then on the interim shelf life of a maximum of 2(x) months may be permitted, where 2(x) does not exceed 3 years.
ii. Shelf lives of longer than 3 years should be supported by data on production batches stored at the maximum recommended temperature for the duration of the proposed shelf life.
iii. Stability trials involving at least two production batches stored at the maximum recommended temperature should be continued for the full period to validate the predicted shelf life.
Interim shelf lives may be extended through the submission of additional data accumulated in the later stages of the trial.
How to calculate shelf life?
After a stability study is completed, shelf life can be predicted from the data by the following methods:
– Statistical analysis, unless obviously within limits
– If analysis shows small batch-to-batch variability, combine all batches into one overall estimate (use p=0.25 to test variation)
– Regression analysis and 95% lower confidence limit
– Transform data into linear relationships, if needed
– Goodness-of-fit on individual and combined methods to test assumptions of linear models

Conclusion
Stability testing is the only way to ensure that pharmaceutical products remain safe and effective throughout their shelf life if kept under recommended storage conditions.
A stability trial requires the manufacturer to simulate real-time and accelerated storage scenarios, store the medicinal products under those conditions throughout their shelf lives, and conduct testing at specified time points (initial, 3 months, 6 months, 12 months, etc.).
This stability testing method helps manufacturers assess the impact of temperature, humidity, and light on the product’s potency and identify potential breakdowns of active pharmaceutical ingredients.
Adherence to rigorous testing regimes, including time-point monitoring and comprehensive assessments of physical, chemical, and microbiological attributes, underpins the reliability of stability studies.
However, the accuracy and validity of results depend on precise trial design, appropriate test methodologies, and vigilant monitoring and reporting.

Author: Kazi Hasan
Kazi is a seasoned pharmaceutical industry professional with over 20 years of experience specializing in production operations, quality management, and process validation.
Kazi has worked with several global pharmaceutical companies to streamline production processes, ensure product quality, and validate operations complying with international regulatory standards and best practices.
Kazi holds several pharmaceutical industry certifications including post-graduate degrees in Engineering Management and Business Administration.