Validation master plan (VMP) – when and how to create one?
- Kazi
- Last modified: March 13, 2025
Have you ever wondered why the regulatory inspectors are naturally keen to examine your validation master plan?
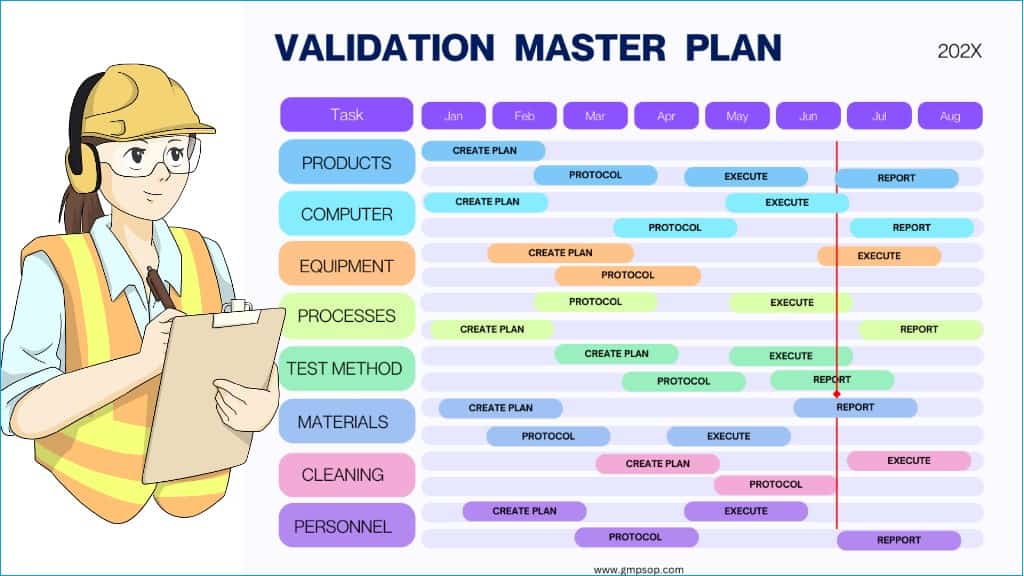
That’s because a well-crafted validation master plan gives them a high-level view of your site validation activities, for example, products, processes, computer systems, equipment cleaning, facility, utilities, etc.
A validation master plan (VMP) outlines the framework for validating critical items, such as in the example above, over a set period (e.g., 12 or 24 months).
It specifies which elements require validation, details the schedules, applicable standards, and assigns responsibilities conducting the validation work.
The plan also helps them understand the overall product risks, how you have identified their priorities, and what validation activities you have scheduled to mitigate the risks.
The validation plan, execution, and evidential records help the regulators build confidence in your product, processes, and systems.
If you are in the pharmaceutical business, regulatory authorities mandate that you develop, implement, and periodically review a validation master plan for qualifying your systems, processes, cleaning method, testing methodologies, equipment, facilities, etc.
This article can help you understand who is responsible for preparing the validation master plan, the stages of the validation life cycle, the risk-based prioritization of validation items, how to prepare a validation schedule, and some practical examples.
Table of Contents
Key Takeaways
- A validation master plan (VMP) is a strategic document that outlines the schedule based on risk, validation approach, and activities, including documentation.
- Life-saving medicines require complex processes, systems, and equipment that must be validated to prove their robustness. The validation master plan ensures that all validation and qualification activities are conducted as required and compliant with GMP guidelines.
- Pharmaceutical validation program should cover new and existing products and all major operational changes.
- FDA regulations (21 CFR parts 210 and 211) ensure companies follow cGMP guidelines in preparing and committing validation master plans. These regulations cover process validation, sampling, testing, and facility qualifications to ensure quality products.
- The five stages of the validation program life cycle begin with the Development and design of the process. Verification that the systems are correctly installed and functional, followed by Qualification to ensure consistent product quality. Commercial production maintains the validated status. Finally, Retirement removes outdated systems.
- Validation master plan examples include process validation, cleaning validation, method validation, computer systems validation and equipment qualification.
- Management of validation items and studies is a key element. This includes products, systems, equipment, or facilities. Validation risk management is another big element, justifying the schedule based on risk.
- Establish validation priorities through product types and modes of use. Like the products and processes, perform risk assessments for equipment and systems by evaluating the level of impact such items will have on them.
- A validation protocol outlines the scope, objectives, and approach to validation, including project description, responsibilities, validation strategy, document management, and acceptance criteria. It should also include flow diagrams and timelines to provide a clear and structured overview that is ready for regulatory review.
250 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents are included each month. All written and updated by GMP experts. Check out sample previews. Access to exclusive content for an affordable fee.
What is a validation master plan (VMP)?
A validation master plan (VMP) is a strategic document that identifies the elements to validate, the approach to each element, organizational responsibilities, and a strategy for maintaining validation documentation.
A validation master plan provides a holistic view of all validation activities your site should engage in for a set period (e.g., 12 or 24 months).
The VMP should provide information on:
– Which items are subject to validation or qualification?
– Schedules of validation.
– Appropriate standards and guidelines to be referenced.
– Functional responsibilities concerning the validation effort.
– Preparation and execution of validation protocol and reporting responsibilities.
– General guidance for validation document format.
The plan should show how you will organize separate validation activities, some of which may be linked internally.
The plan should provide the details and relative timescales for the validation work that needs to be performed.
It provides a framework for consistently validating all critical processes, equipment, and systems used to produce products of the desired quality and safety standards.
In summary, the validation master plan is a foundation for all validation activities within your facility.
Validation master plan for the pharmaceutical industry?
The pharmaceutical industry manufactures life-saving therapeutic products. It uses sophisticated processes, systems, and equipment to turn starting materials into finished medicines.
These facilities bear the burden of proof and must generate evidence that their product is safe and pure.
Regulatory authorities require them to produce evidence that the processes and systems are validated, critical equipment is qualified, and manufactured products are tested for purity.
That is not the end of their responsibility. Pharmaceutical companies are accountable for properly warehousing, shipping their products, and guaranteeing their stability until expiry.
The validation master plan provides the first line of defense that all required validation and qualification activities are performed on a schedule and that their processes are compliant with GMP guidelines.
The validation plan applies to all existing and new drug compounds and registered drug products or active pharmaceutical ingredients (APIs) for clinical use or sale.
The validation master plan in the pharmaceutical industry must provide evidence that the following major changes comply with regulatory expectations.
– The receipt and establishment of new drug products or API’s.
– Major processing changes to existing drug products or API’s.
– The construction of new manufacturing or related facilities.
– Major alterations to existing manufacturing or related activities.
Regulatory requirements for validation plan
Suppose you are in a business that manufactures and sells medicinal products for human use. In that case, your products must be registered with your local regulatory authority with sufficient evidence that they are safe, pure, effective, and traceable.
It is a regulatory requirement that you provide evidence-based justifications showing that the validation and qualification stages you have undertaken are sufficient to ensure that your processes consistently produce a product of the desired quality.
According to the FDA, process validation for drugs, including both finished pharmaceuticals and their components, is mandated by law under section 501(a)(2)(B) of the Federal Food, Drug, and Cosmetic Act.
This mandate requires adherence to current good manufacturing practice (CGMP) regulations outlined in 21 CFR parts 210 and 211.
cGMP regulations necessitate that manufacturing processes be planned and monitored to ensure consistency and reliability in meeting quality standards.
As specified in § 211.100(a), process validation requires written production and process control procedures to consistently meet product attributes such as identity, strength, quality, and purity.
Other CGMP regulations guide the validation process, such as:
i. Sampling and testing of in-process materials,
ii. Establishment of in-process specifications consistent with final product specifications and
iii. Ongoing product quality and manufacturing experience review to determine necessary process changes.
iv. Facility and equipment requirements are specified to ensure proper operations and performance.
In summary, cGMP regulations ensure that manufacturing processes consistently and reliably produce products that meet quality requirements.
When is a validation master plan required?
A validation master plan is needed before a new product, process, and system is commissioned at your facility.
You would also require a master plan when you make significant changes to the facilities, equipment, and processes that may affect the quality of the product.
For projects involving major changes to existing equipment, a new validation master plan should be prepared and available before starting any validation activities.
To determine the scope and extent of validation, you must use a risk-based approach. The validation master plan should be available before starting any validation activity.
A VMP is not required for projects that involve installing or altering a single piece of equipment. Document these tasks on separate validation plans and reports.
Sometimes, a site-specific VMP (for the site’s validation requirements) and project-specific plans may exist simultaneously.
Who is responsible for writing a validation master plan?
Your site should have standard operating procedures (SOPs) detailing who should be responsible for developing validation master plans, who should approve the plan, who should execute it, and how to refer individual validation projects to the master plan.
The Technical Services or Validation team should develop all validation plans, and quality assurance is typically responsible for approving them and related documentation.
The validation manager should identify the stakeholders and assign the subject matter expert responsible for validation activities. The manager should also provide carefully constructed protocols and training to perform such activities while overseeing all aspects of a validation project.
 – when and how to create one? 1")
The validation master plan should be reviewed, commented on, and approved by the facility’s senior management or a steering committee comprising subject matter experts.
Where consultants are used to preparing plans, their work should be subject to the same level of scrutiny and approval as in-house documentation.
When third-party contractors are used to manufacture an intermediate or API, the quality assurance team of the lead site is responsible for ensuring that the contractor’s facilities, equipment, and processes are qualified and validated as per site requirements.
250 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents are included each month. All written and updated by GMP experts. Check out sample previews. Access to exclusive content for an affordable fee.
How to create a validation master plan template?
You should prepare a validation master plan using templates that cater to each of your facility’s critical processes and systems and have a schedule for each type of validation: process, cleaning, computer system, and method validation.
These templates can differ greatly due to their inherent differences in functionality. However, some common items to include in the template are the scope of work, the objective of the plan, detailed validation requirements, and validation approaches. Include the list of products, processes, systems, equipment, and personnel who should be involved.
Include the key project details, such as project name, number, equipment specifications, process location, and schedule of activities in time and date.
Specify the responsibilities, such as the personnel executing the validation activities and those reviewing and approving the template.
I have included here an example of validation master plan template for your reference.
05 stages of the validation program life cycle?
The validation program looks at every step of a system’s life to ensure that important processes, systems, and equipment are well-made, installed, and tested.
They should also be always maintained and used according to the GMP rules.
When a critical system, process, or product becomes too old and ineffective, it will be taken out of service.
 – when and how to create one? 2")
Stage 1: Development and design (building and capturing process knowledge and understanding):
i. At this stage, perform activities related to designing and developing processes, process trains, and critical systems. Generally, this stage does not occur under Good Manufacturing Practice (GMP) conditions.
However, you should follow sound scientific methods, principles, and good documentation practices at this stage.
ii. The primary objective of this stage is to establish commercial process and operation requirements based on knowledge gained from design, development, and scale-up studies.
This includes identifying process parameters and intended procedures, understanding control and quality attributes, and considering potential sources of variation in commercial processes and systems.
iii. Ensure that process development, pilot work, and commissioning provide supportive evidence for commercial operation verifications and process qualifications.
This will help in validating the commercial processes effectively.
Stage 2: Verification
i. At this stage, activities will be undertaken to provide evidence of the successful installation and operation of critical equipment, process trains, and systems affecting product quality and efficacy.
Verification studies are conducted under good manufacturing practice (GMP) conditions.
ii. The primary goal of this stage is to confirm that systems are installed and operated as designed during Stage 1.
This includes verifying design principles and operational procedures and developing calibration and maintenance systems to ensure ongoing compliance.
iii. Conduct verifications before undertaking Stage 3 studies.
This ensures that systems are properly validated before proceeding to the next stage of the validation life cycle.
Stage 3: Qualification and validation
i. This stage encompasses all activities demonstrating that a process, product, system, or critical train can consistently produce products that meet safety and quality standards.
ii. Adopt an “A day in the life” approach, conducting testing within normal operating conditions and using commercial manufacturing methods. Collect and evaluate data during each study to ensure consistency and safeguard against batch variation.
iii. Use knowledge from the design, development, and verification stages to establish parameters such as critical control points (CCP) and specifications like critical quality attributes (CQA).
In this stage, verify or validate these parameters and specifications.
iv. Consider shortening activities at this stage if sufficient development and trial batch studies have been conducted.
Document justification for reducing validation requirements, demonstrating that prior studies were performed under GMP or near-GMP conditions and met quality attributes associated with commercially released products.
v. Develop a protocol and report in line with the VMP requirements when utilizing design and development studies. Demonstrate process proficiency and technical mastery throughout these activities.
Stage 4: Commercial production
i. Upon completing the initial validation stages, release the product, process, equipment, or system for routine production.
This stage may involve further testing at the same level as stage 3 qualification, especially for concurrently produced batches, or increased testing until sufficient data is collected.
ii. The goal is to maintain the process, critical equipment, and systems in a state of control (validated state) during routine production.
Develop a system that detects deviations from accepted standards through evaluation and review processes.
iii. When you detect a deviation from the standard, assess the impact and define necessary activities to effectively manage and control the variation’s cause.
This ensures ongoing quality and consistency in commercial production.
Stage 5 – Retirement
The final stage of the product validation lifecycle is the retirement of the process and system once they are no longer in use. Such retirements will be documented and controlled through the change control system.
240 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents included each month. All written and updated by GMP experts. Checkout sample previews. Access to exclusive content for an affordable fee.
Validation master plan examples
The validation master plan works as a navigational tool to answer what needs to be validated, when to validate, to what extent, and when to revalidate.
The plan must be clear and not ambiguous about the performance of a process, system, or equipment.
Following are some examples of validation studies which should be included in the validation master plan:
1. Process validation master plan
The process validation master plan accounts for all manufacturing and packaging processes directly employed in the manufacturing and packaging registered products.
The process validation of formulated products (e.g., tablets, capsules, ointments, etc.) starts with a well-defined validation plan and protocol.
Process validation refers to testing the reliability of formulated products under pre-determined processing conditions.
It begins by identifying critical process steps and parameters (e.g., blending, granulating, tableting, etc.), conducting validation risk assessment, and considering potential adverse consequences if they are not met.
The validation protocol outlines the steps to ensure the process consistently produces a formulated product meeting quality standards.
Essential steps you should take during process validation activities are:
– Develop a validation master plan.
– Prepare validation protocol.
– Identify prerequisites for process validation.
– Select validation approach
– Worst-case and challenge tests
– Determine matrixing or family approach
– Perform validation testing
– Validation Documentation
– Manage changes
If you need more guidance on preparing a master plan, refer to the Preparation of a validation master plan and Master Validation Plan.
2. Cleaning validation master plan
The goal of the cleaning validation master plan is to ensure that all the facilities and equipment are accounted for.
The plan ensures every equipment is clean and free from contamination, which can impact the quality of your products.
Cleaning is the removal of visible and microscopic contamination by dirt, extraneous matter, or product residues by mechanical or physical means.
The purpose of cleaning validation is to provide documented evidence that the cleaning processes used in a manufacturing facility effectively remove residues from a previous batch, cleaning agents, or microbial contamination below pre-established maximum allowable carryover limits.
You can read our article “Equipment Cleaning in Pharmaceutical” to explore cleaning validation further.
If you need help with complete guidance, please refer to the cleaning validation master plan.
3. Method validation master plan?
Analytical method validation, also called methodology or test method validation, involves demonstrating that the analytical methods used to test the quality of your product are accurate, reliable, and reproducible.
This is critical for ensuring the quality and safety of your products.
While creating a method validation master plan, coordinate with the quality control and formulation teams to identify all methods employed to test medicinal products in the laboratory.
Create a master plan listing all products, specifications, and test methods. Validate those test methods using the steps below:
– Identification and evaluation of the critical parameters of the analytical methods,
– Perform method validation studies,
– Document & evaluate the results.
To complete an analytical method validation, you will need to validate all or a combination of the following parameters:
– Precision, repeatability, intermediate precision.
– Method accuracy
– Sensitivity: Limit of detection (LOD)
– Sensitivity: Limit of quantitation (LOQ)
– Selectivity/specificity and system suitability
– Method linearity and range
– Method ruggedness and robustness
If you need help with complete guidance, please refer to the method validation master plan.
250 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents are included each month. All written and updated by GMP experts. Check out sample previews. Access to exclusive content for an affordable fee.
4. Computer systems validation plan
Computer system validation ensures that all computerized systems (including hardware and software) that directly impact product quality operate as per the required specifications and principles of GxP.
The computer validation plan lists all computerized systems directly employed in the manufacturing and testing of products.
Complete a risk-based categorization of those systems between the direct and indirect impact on the processes and products.
You should be able to prepare the validation plan, which will outline the principles and objectives of your facility’s computer validation program.
You should conduct all validation activities according to the schedule in the master plan. The plan will provide an overall picture of all validation activities to ensure that computer systems are sufficiently qualified to support product quality and safety.
Validation of computer systems provides documented evidence to assure that they will consistently function according to their pre-determined specifications and quality attributes throughout their lifecycle.
Important aspects of computer system validation are:
– The formal management of design (through a specification process).
– System quality (through systematic review and testing).
– Risk (through identification and assessment of novelty and critical functionality).
– Lifecycle (through sustained change control).
Where equipment is controlled by embedded computer systems, computer validation elements may be performed as part of the equipment installation qualification and operational qualification protocols.
If you need more guidance, you can refer to the Computer systems validation master plan.
5. Equipment qualification plan
Equipment and facility qualification, an integral component of the validation master plan, is pivotal in ensuring the quality and safety of manufacturing processes.
Successful qualifications prove that the equipment, facilities, and utilities you use to manufacture medicinal products perform well and meet the required quality standards.
The extent of equipment qualification should be based on risk assessment.
The validation plan should list all critical aspects of equipment, function, utilities, and other system components.
The plan should include a timeline for the qualification and verification of all direct impact components to generate evidence that they are under control.
The risk assessment should be based on the impact on product quality and patient safety.
Additional items for consideration include
– The level of standardization,
– Level of complexity,
– Configuration,
– Customisation,
– Intended use, and
– Vendor quality system assessment where applicable.
Equipment and facility validation plans should include periodic reviews of equipment validation status that come in contact with medicinal goods during production.
Validation master plan flow chart
We have drawn a simple flowchart below to give you an idea of what is involved in creating a validation master plan and how you would execute it.
First, you should identify the validation items based on impact and risk, followed by an individual validation plan, create validation protocols, assign a team to execute the validation studies and prepare final reports.
 – when and how to create one? 3")
Key elements to consider during validation activities
We have told you the common validation examples typically undertaken by the pharmaceutical manufacturing facility. Also, we have demonstrated the five stages of the validation lifecycle.
The next step is to assess some special factors and considerations to help you prepare your validation master plans.
1. Management of validation items and validation studies
Validation Items are physical products, systems, equipment, or equipment trains that are product-related that can significantly impact product quality or safety.
Validation item records are stored in the designated validation database and enable you to track the validation status of those items.
Validation item records are living documents where you can monitor the current status, planned and completion dates, area (location), item, and types of validation.
Validation studies ensure that physical products, systems, and equipment that directly impact product quality are implemented as designed, compliant with Good Manufacturing Practices (GMP), and suitable for their intended purpose.
The extent of validation studies depends on the stage of the development life cycle and the perceived risk level of the item being validated.
Assign a unique project number for each validation study and track it within a validation database.
Typically, a validation project involves tasks or actions taken to ensure the validity of one or multiple physical items.
2. Validation risk management
The scheduling of a validation master plan largely depends on the risk profile of the validation items,
Appropriate categorization of risks will dictate the validation priorities and successively aid in preparing schedules with timelines.
By default, you should prioritize validating new products, processes, equipment, facilities, and systems once they are proven highly critical through risk assessment.
3. Establish validation priorities for products and processes
Overall, the idea is first to identify the product types and mode of use and then evaluate the criticality of processes and products to assess their risk base.
Once identified, you should include these processes or products in the validation master plan and validate them according to risk priority.
We have prepared the following example of a risk-assessed prioritization table based on product types and modes of usage.
Develop your table of priorities, which will help you understand the risks to products, schedule validation activities in the plan, and mitigate perceived risks.
 – when and how to create one? 4")
250 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents are included each month. All written and updated by GMP experts. Check out sample previews. Access to exclusive content for an affordable fee.
4. Establish validation priorities for equipment and systems
Like the products and processes, perform risk assessments for equipment and systems by evaluating the level of impact such items will have on products and processes.
The impact assessment of equipment and systems to be validated can be divided into three categories based on the following criteria:
High Risk – direct impact equipment
An equipment or a system is defined as “Direct Impact” if it meets any of the following criteria:
– The system has direct contact with the product.
– The system provides an excipient, ingredient, or solvent.
– The system is used in cleaning or sterilization.
– The system preserves product status.
– The system produces data used to accept/reject products.
– The system is a process control system.
Medium Risk – indirect impact equipment
Indirect impact equipment is equipment where a process deviation involving the equipment will impact the product quality.
However, those deviations can likely be easily detected through in-process or final QC testing.
Examples of such equipment are:
– Laboratory Freezers and Fridges,
– Cold Rooms
– Incubators
– Water baths
– Power Packs
– Label Printers
Low Risk – no impact equipment
The equipment or service does not directly or indirectly impact product quality or safety.
In this instance, the equipment or process does not require ongoing performance monitoring and does not impact quality-critical performance claims.
In-process or final pack testing, for example, non-product contact utilities and component handling equipment, will have a high probability of poor performance detection.
Once you can define the inherent risks of products, processes, systems, or equipment, you can develop the validation master plan based on the priority of validation activities that can effectively control and mitigate those risks.
Follow the risk management process flow as shown in the following figure:
 – when and how to create one? 5")
5. Determine validation Vs. verification points
While developing your master plan, please remember not all critical steps in a process or system require validation or qualification.
Sometimes, controlling the risks through routine verifications and monitoring is more appropriate wherever possible.
This should reduce the overall burden on the resources without compromising the necessity of validation or overlooking the obvious.
If you are faced with a dilemma between validation or verification points, use the following decision chart to document your assessment and outcome.
250 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents are included each month. All written and updated by GMP experts. Check out sample previews. Access to exclusive content for an affordable fee.
 – when and how to create one? 6")
How do you prepare validation protocol for VMP?
The content of the validation master plan should reflect the complexity and extent of the validation activities to be undertaken.
At a minimum, you should include the following items in a validation protocol.
– Title, statement of commitment, and approval page.
– Summary description of the project and its scope.
– A statement of validation policy and the objectives of the validation activity.
– References to other existing validation documents.
– A description of the organization and responsibilities for validation.
– The validation strategy you will adopt for facilities and systems (e.g., process equipment and automated systems), materials, quality control, and personnel, including training.
– The intent concerning process validation and cleaning validation for each drug product range.
– The document management and document change control system to be used.
– A description of the validation change management process.
– An indicative relative timescale plan.
– Clear acceptance criteria against which the outcome of the validation exercise will be judged.
You can find an example of a validation master plan protocol from this link.
Each protocol describes the scope of the activities and addresses relevant key elements of validation, indicating the actions to be taken and documents that will be needed.
The key elements are those factors that can affect product quality.
The protocol should identify all the components to be included within a validation project.
You should include a flow diagram or matrix that can be very useful as an overview and monitoring tool. You should also include a high-level process map or flowchart of the manufacturing process.
A materials validation plan may be used for large projects involving many materials. It is optional for smaller projects. The plan or lower-tier documentation alone may cover the qualification of materials.
The validation plan should demonstrate that the validation activities have been considered and are being organized in a structured manner.
You should prepare a validation master plan to be presentable as a formal document suitable both as an internal guide and for scrutiny by a member of regulatory inspectors.
Therefore, it should be concise, easy to read, and not excessively duplicate text from documents held elsewhere in the quality system.
Documentation and record-keeping in a master validation plan
A validation master plan is an essential part of the overall validation program for a pharmaceutical site. The documentation and records-keeping process for the validation program should reflect this.
We can classify the validation documentation into two major segments.
1. Validation as a system documentation
Validation as a system document is defined as those documents describing the management of a validation system or those documents created to guide critical tasks.
You should list the validation system documentation as an appendix of the validation master plan.
You should establish the following system documents in the validation program:
i. Validation master plan (VMP), review every 3 years.
ii. The validation master schedule (VMS) is reviewed yearly.
iii. The product master list for each site is reviewed yearly.
iv. The equipment and systems master list for each site is reviewed yearly.
v. Validation Guidance (SOP)
2. Validation documents for evidence
Validation documents for evidence are those documents that provide a level of assurance that a system has been designed, installed, and operated correctly and is fit for its intended use.
These documents are generated throughout the validation life cycle as required.
a. Development and design documents.
These documents provide an overview of how the processes, systems, equipment, and facilities are developed and designed that are used on the site.
There are five types of evidence-based design documentation.
– User requirement specification (URS)
– Design specification (DS)
– Functional design specification (FDS)
– Product design specifications (Technical Transfer)
– Development trials and studies
b. Verification and qualification documents
These documents provide direct assurance that a given process operates within defined critical process parameters and has met its critical quality attributes (CQA).
These documents are:
– Installation qualification (IQ)
– Operational qualification (OQ)
– Equipment verification (EV)
– Performance qualification (PQ)
– Product validation (PV)
– Aseptic operations validation (MV)
– Computer system validation (CSV)
– Revalidation (R)
– Retrospective review (RR)
– Validation investigation or study (VS)
– Validation report (VR)
250 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents are included each month. All written and updated by GMP experts. Check out sample previews. Access to exclusive content for an affordable fee.
Conclusion
A validation master plan (VMP) is a high-level document that captures all the validation items, risk-based prioritization, schedule of validation activities, and documentation that assures your processes and systems are working as intended.
Regulatory inspectors are keen to examine your validation plans because the plan provides a comprehensive overview of your validation programs and activities in a single document.
Regulatory authorities require pharmaceutical companies to develop, execute, and periodically review their validation master plan to ensure the validation and qualification of systems, processes, cleaning and testing methods, equipment, and facilities are under control.
The validation master plan covers the validation activities before a new product or process is introduced. It should also cover once major changes impact them.
Before developing a master plan, you need to understand the different stages of the validation life cycle, including design and development, verification, qualification, commercial use, and retirement of validation items.
A typical validation workflow covers all areas under validation, including creating individual plans and protocols, executing them, and producing reports.
Risk management is vital to evaluating priorities, such as identifying direct vs. indirect impact processes and systems to put under validation first. Sometimes, risks can be well managed through in-process verification instead of full-blown validation activities.
To avoid ambiguity, you need to select your validation or verification activities through expert assessments and document all rationales in your validation master plan.
Lastly, there is no alternative or excuse for not having a well-documented validation program. Regulators love these records. Moreover, well-managed records are a testament to your effort to make your products safe.
 – when and how to create one? 7")
Author: Kazi Hasan
Kazi is a seasoned pharmaceutical industry professional with over 20 years of experience specializing in production operations, quality management, and process validation.
Kazi has worked with several global pharmaceutical companies to streamline production processes, ensure product quality, and validate operations complying with international regulatory standards and best practices.
Kazi holds several pharmaceutical industry certifications including post-graduate degrees in Engineering Management and Business Administration.